
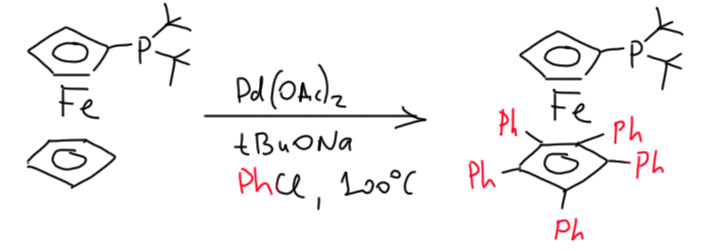
CH-активация
Реакции органических соединений чаще всего происходят на определенных реакционных центрах, атомах, несущих уходящие группы, включенных в кратные связи и т.п. Но больше всего в органических соединениях атомов водорода, связанных с атомами углерода. Это настолько банальная и вездесущая часть структур органических соединений, что мы обычно вообще их не замечаем, никак не обозначаем их в структурных формулах, – просто и совершенно инстинктивно мысленно добавляем атомы водорода для соблюдения правильных валентностей.
Тем не менее, именно атомы водорода на углеродах – самые лакомые кусочки органической структуры для органиков, разрабатывающих новые методы синтеза. Если удается целенаправленно и более-менее селективно заменить атом водорода на какую-нибудь группу или фрагмент, радости не бывает пределов, и самые гламурные научные журналы с радостью распахивают свои страницы для таких сообщений и статей. Почему? Строго говоря, потому, что это, во-первых, очень модно, и всё тут, а во-вторых, все знают, что это очень модно. Это главное. Научная жизнь в целом устроена точно так же, как и жизнь мира моды и шоу-бизнеса. Объяснить, почему что-то сейчас в тренде, на обложках, в речах хедлайнеров престижных конференций, а что-то украсило собой задворки жизни, практически невозможно. Мы вот только в предыдущей лекции потешались над другой гламурной темой – реакциями углекислого газа в контексте проблем с глобальным потеплением.
Но наука, безусловно, всегда старается для любого тренда придумать рациональное обоснование. В случае с атомами водорода в ход идет, в первую очередь, спасительный принцип экономии атомов. По крайней мере, с точки зрения исходных веществ и продуктов замена атомов водорода на что-то важное и полезное более всего удовлетворяет этому принципу из всех реакций замещения (реакции присоединения в любом случае вне конкуренции) – так как замещается, уходит в помойку, самый легкий из всех известных атомов, атом водорода. В этом месте обязательно нужно сделать вид, что мы не знаем, что атомы водорода сами по себе на помойку не отправляются, а непременно прихватывают еще что-нибудь – кислотный остаток, основание, акцептор атома водорода, из того, что было в реакционной смеси помимо исходного органического субстрата, и масса этого довеска будет в десятки, сотни, а то и тысячи раз превосходить массу атома водорода. Но умение деликатно делать вид, что мы не замечаем что-то такое, что замечать в данном контексте не положено – важнейший признак образованного, культурного человека. Так же и в мире моды – мы же не спрашиваем, отчего вон та модель надела на голову ведро. Если пришли на модный показ, подразумевается, что знаете, отчего можно надеть на голову ведро. А если не знаете и позволяете себе мерзко хихикать – прочь с модного показа, невежда!
Но если еще немного подумать, то придется признать, что доля правды в представлении об “атомной экономичности” методов, связанных с CH-активацией, все же есть. Если в начале всех синтетических цепочек стоят самые простые исходные, то для того чтобы ввести в молекулы хорошие уходящие группы или другие реакционные центры приходится использовать всякие вспомогательные реакции (галогенирование, нуклеофильное замещение и т.п.), часто с введением и снятием защитных групп, и цена превращения сильно возрастает, а количество отходов увеличивается. И если прикинуть всё, что пошло в помойку в метода, использующем CH-активацию, и в методе, использующем другие уходящие группы, то часто окажется, что CH-активация действительно экономнее, и даёт меньше отходов, даже если скрупулёзно посчитать всё. когда и правда проще и экономичнее придумать метод прямой модификации исходных по CH-связям. Реальность всегда сложнее принципов и идей, поэтому примем простую мысль, что запас карман не тянет, и лишние методы никогда не помешают. В лабораторном синтезе, да и в тонком органическом синтезе ценных соединений возможность получить продукт всегда важнее пути, которым этот синтез осуществляется.
Итак, мы хотим использовать в реакциях в качестве реакционных центров самые обычные CH-связи. Подразумевается, что обычно они недоступны или труднодоступны для реакций. Дело здесь часто бывает вовсе не в низкой реакционной способности таких связей, а скорее в том, что в органических молекулах полно разных связей этого типа, и задача целенаправленно выбрать только какие-то конкретные может на первый взгляд показаться вообще нерешаемой. Но она решается, и весьма типичным для органической химии способом. Нужно найти побольше методов, позволяющих воздействовать на связи нужного типа. У каждого из таких методов будут свои требования к структуре, электронным эффектам, влиянию соседних групп, стерическим факторам. Если мы располагаем большим набором методов, то в том случае, когда нам понадобится реально добраться до какого-то конкретного атома водорода в конкретной молекуле, есть надежда, что минимум один из коллекции методов подойдет. И тогда мы прославим подходящих богов и вождей, и скажем, что именно так и было задумано.
.
В органической химии реакции CH-связей очень распространены во всех типах органических соединений. Воздействие на CH-связь достигается одним из трех хорошо известных основных способов, хотя есть ещё и другие. Свободнорадикальная активация применима к CH-связям на насыщенном атоме углерода и заключается в отрыве атома водорода радикальной частицей, с последующим превращением углеродного радикала. Электрофильный способ уже применим и к насыщенным, и к ненасыщенным, в основном ароматическим фрагментам и заключается в образовании реакционноспособных карбениевых и карбониевых ионов, и их реакциям. Нуклеофильная активация наиболее хорошо разработана и применяется к CH-связям на всех трех основных типах углеродных атомов: она заключается в отрыве протона от фрагментов с достаточно высокой CH-кислотностью с последующим превращением карбаниона.
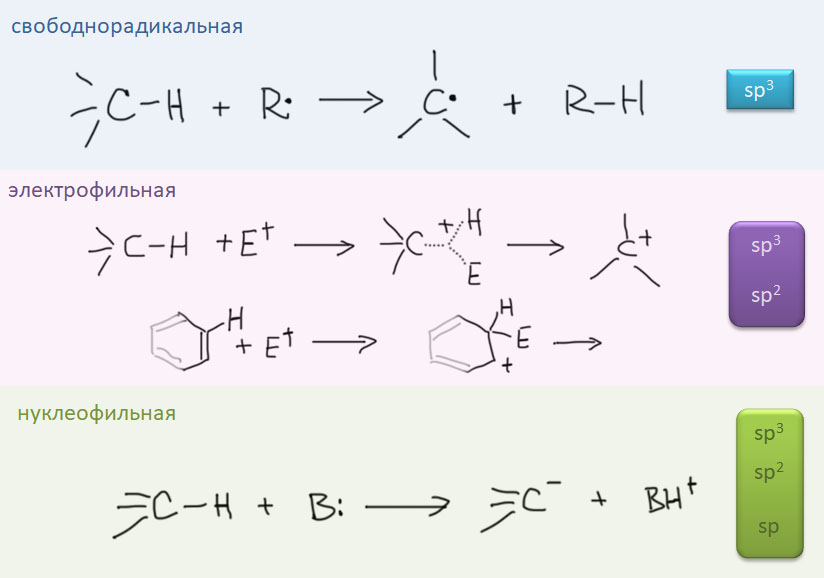
Возникает логичный вопрос - если обычная органика так хорошо умеет это делать, зачем тут переходные металлы? А всё та же экономия атомов - в обычной органической химии ведь мы сначала рвём связь C-H какими-то реагентами, и только после превращаем интермедиаты в продукты. Переходные металлы могли бы нам позводить соединить эти два действия в одном процессе, совсем хорошо если в каталитическом цикле.

Самая сложная задача – активация алифатических CH-связей, особенно в простых алканах. Для лабораторного и тонкого органического синтеза эта задача малоактуальна, но для крупнотоннажной промышленной химии алканы – наиболее подходящее сырье, и прямая переработка алканов в ценные полупродукты – задача чрезвычайно важная просто по экономическим соображениям. На исследования возможных путей селективной активации алканов тратились и тратятся колоссальные средства. Успехов пока не очень много. Может быть, эта проблема вообще не имеет решений? Может быть, но все же некоторую надежду дает и то, что с этой задачей довольно легко справляются живые организмы, в том числе и с помощью металлокомплексных ферментных систем, и то, что за долгие годы исследований было открыто несколько перспективных процессов, показывающих, что принципиальная возможность использования алканов в крупном синтезе есть. Безусловно нельзя забывать про гетерогенный газофазный катализ – дегидрирование алканов в алкены давно уже является источником олефинов для промышленности, хотя самые востребованные олефины – этилен и пропилен – в основном получают некаталитическим паровым крекингом алканов – свободнорадикальной реакцией при высокой температуре. Но мы здесь обсуждаем гомогенные процессы в жидкой фазе. Принципиальную разницу между гетерогенным катализом и гомогенным (в том числе и гетерогенизованным гомогенным) катализом мы уже обсуждали.
Основополагающее открытие в активации алканов комплексами переходных металлов было сделано в 1972 году Шиловым и сотр, показавшим, что метан и другие алканы превращаются в спирты и хлорпроизводные в каталитической системе на основе комплексов платины. Каталитическая система требует участия окислителя для перевода Pt(2+) в платину (4+), и в оригинальном процессе Шилова участвовали комплексы обеих степеней окисления. Активация C-H связи достигается за счет электрофильного расщепления, при котором водород уходит в виде протона. Образование продукта реакции осуществляется за счет SN2-замещения в комплексе Pt(4+), причем платина с остальными лигандами становится уходящей группой. Именно окисление Pt(2+) в Pt(4+) и создает хорошую уходящую группу для замещения.
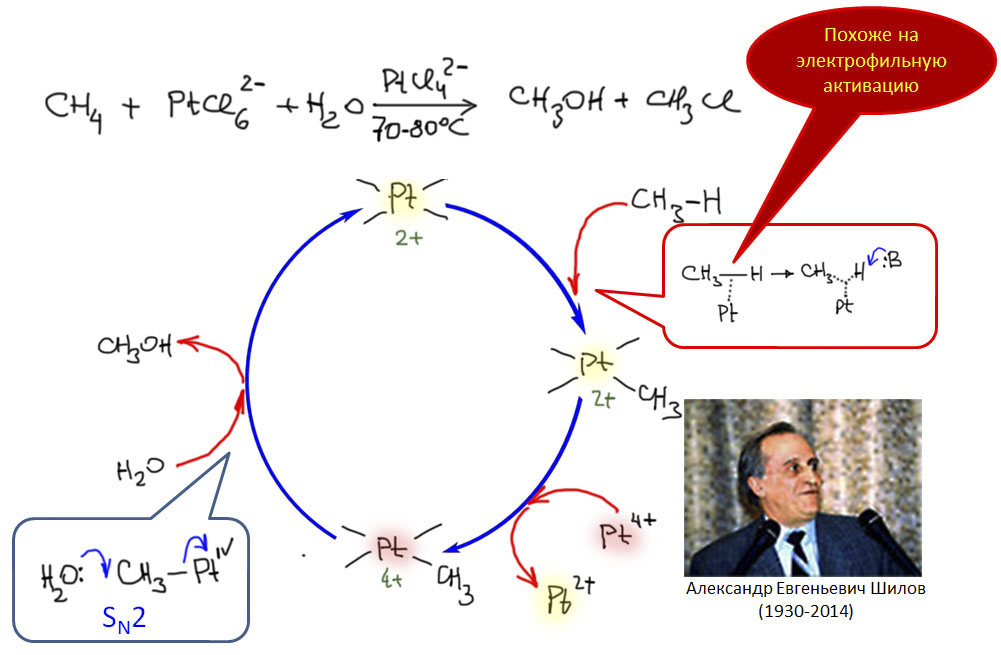
Реакция Шилова дает ключ к разработке процессов превращения алканов в полезные продукты, но сама по себе характеризуется низкими значениями TON и TOF, и не имеет практического значения. К сожалению, где находится дверь, ключ к которой нашёл Шилов с сотрудниками, до сих пор установить не удалось, хотя искали её многие. Предполагалось и отчасти до сих пор предполагается, что за этой дверью находится новая химия (Shilov's chemistry) селективных и мягких превращений алканов, которая перевернёт весь промышленный органический синтез.

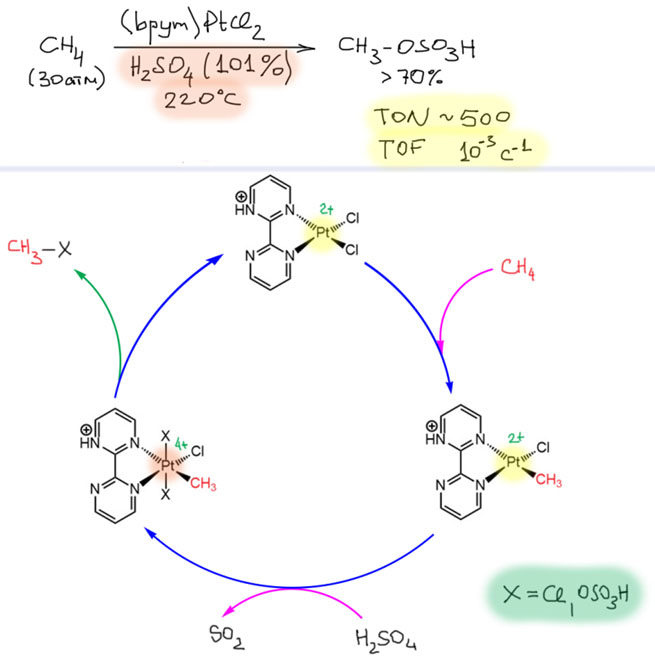
Наиболее эффективная реализация реакции Шилова была получена в 1998 (Periana, R. A. et al., Science 1998, 280, 560) и запатентована компанией Catalytica. Реакция идет в невероятно жестких условиях. Кроме довольно большого TON была решена и вторая проблема - в этой системе не используется соединение платины в качестве стехиометрического окислителя. Реакция идет в горячей серной кислоте (моногидрате), которая и работает как окислитель. Интереснейшей особенностью метода является использование комплекса платины с 2,2'-бипиримидином, который обладает беспрецедентной устойчивостью и сохраняется в среде с огромной кислотностью в монопротонированной форме, что обеспечивает и растворимость комплексов в необычной среде реакции. Основным продуктом реакции в таких адских условиях является метиловый эфир серной кислоты, и это фактически ставит крест на практическом применении метода, так как такой продукт невозможно выделить из раствора в серной кислоте. Очевидно, что практическая эксплуатация каталитического окисления алканов по Шилову требует дальнейших исследований, но за 20 лет, прошедших с открытия процесса Periana-Catalytica, про новые прорывы в этой области ничего не слышно.
Гораздо больших практически значимых успехов удалось достичь в активации CH-связей в ароматических соединениях. Эта область с середины нулевых годов и до сих пор переживает бурный рост. Разработаны многочисленные протоколы кросс- и гомо-сочетания ароматических соединений, аналоги реакции Мидзороки-Хека, карбонилирования и множество других полезных реакций. Разработанные методы уже широко применяются в органическом синтезе. Здесь мы встречаемся с такой важной вещью как палладациклы, которые интересны не только как промежуточные соединения в направленных реакциях, но и сами по себе.
Многие металлы в положительных степенях окисления, в том числе непереходные, обладают способностью электрофильно атаковать ароматические кольца в реакции электрофильного ароматического замещения. Наиболее знаменита такими реакциями ртуть(2+), мощный электрофил. Характерной чертой реакций электрофильного металлирования является не очень высокая позиционная селективность, и с заместителями разного типа получаются смеси продуктов. Эта особенность косвенно свидетельствует о том, что механизм металлирования (меркурирования и проч.) не полностью соответствует классическому механизму ароматического электрофильного замещения, в котором ключевыми интермедиатами являются делокализованные карбокатионы, сигма-комплексы. Этот врождённый недостаток надолго затормозил развитие методов органического синтеза, основанных на реакции электрофильного металлирования.
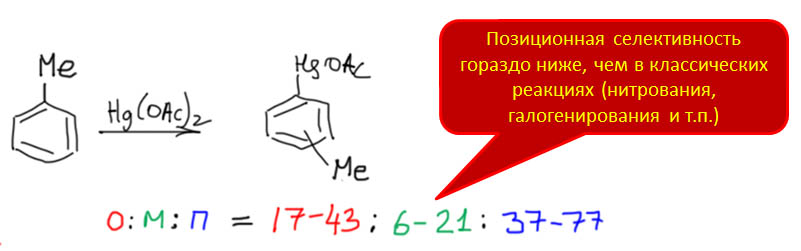
Среди переходных металлов эта способность была давно известная для палладия (2+), который ведет себя очень похоже на Hg(2+). В ранних работах для палладирования всегда использовали более электрофильные простые комплексы палладия типа ацетата или трифторацетата. Позиционная селективность электрофильного палладирования так же низка, как и в реакциях меркурирования, а кроме того, они не так устойчивы в условиях реакции палладирования, а превращаются в другие продукты, в частности дифенилы, но образование сложных смесей изомеров делал эту реакцию непригодной для применения, хотя она известна с 1960-х и время от времени привлекала внимание исследователей.
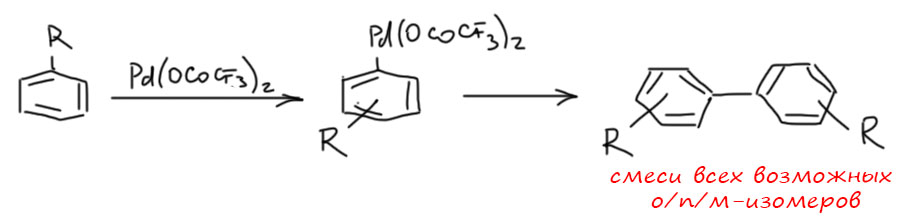
Электрофильное палладирование дает точно такой же арилпалладиевый комплекс, как и окислительное присоединение к арилгалогенидам (с точностью до X-лиганда, который может быть галогенидом или ацетатом или еще каким-то кислотным анионом). Поэтому неудивительно, что такой комплекс может реагировать с олефином точно так же, как в реакции Мидзороки-Хека. Эта реакция была открыта в 1967 г Юдзо Фудзиварой и Итиро Моритани в японской Осаке даже раньше, чем сама реакция Мидзороки-Хека (в 1967). Отличие этой реакции прежде всего в том, что она не каталитическая, а стехиометрическая, а кроме того, обладает низкой позиционной селективностью, поскольку последняя определяется электрофильным палладированием. По этим причинам после появления реакции Мидзороки-Хека эта реакция была фактически забыта до начала нового века.
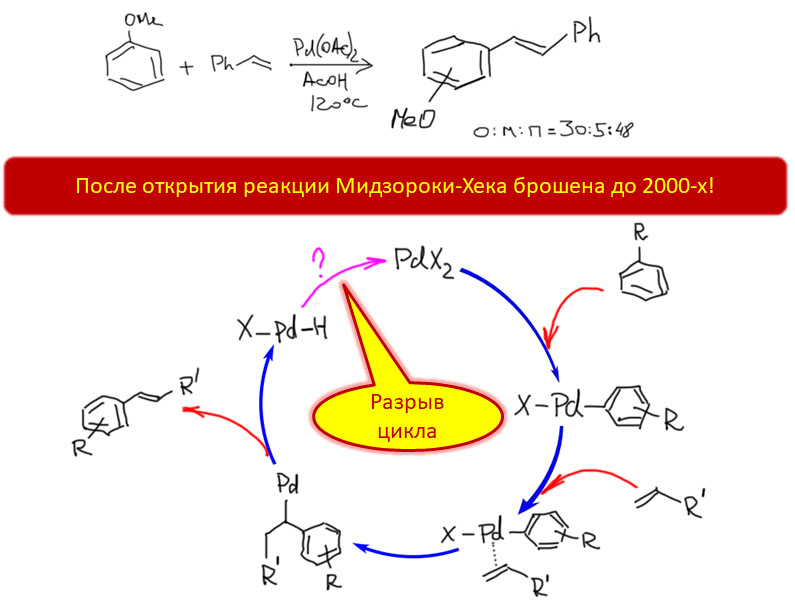
Первооткрыватели считали, что предлагают продолжение Вакер-процесса, то есть рассматривали ароматическое соединение как нуклеофил, присоединяющийся к активированному палладием олефину. Как мы знаем, в реальности всё наоборот, и палладий сначала активирует именно ароматическое соединение, но сама по себе аналогия очень интересная, и лишний раз показывает, как тесно связаны многие реакции. Хотя сами авторы впоследствии смогли предложить и каталитический вариант, использовав точно такой же приём как в Вакере - регенерацию Pd(2+) солями меди или серебра, потребовалось время, чтобы разобраться, что в ней происходит, и как этим можно попробовать управлять.
После появления работ Хека, Фудзивара и Моритани переосмыслили механизм своей реакции, точно поняв ее тесную связь именно с открытием Хека.
Когда мы говорим про обычные электрофильные реакции в ароматическом ряду (нитрование, например), мы видим очень чёткие признаки таких реакций. В первую очередь это влияние заместителей на реакционную способность (активируют или дезактивируют) и ориентацию замещения (орто-пара против мета). И знаем, что от нитробензола до анизола расстояние по реакционной способности чудовищное - сильно больше десяти порядков.
Поэтому никогда не используют один реагент для анизола и для нитробензола (нитрующий, сульфирующий, ацилирующий и т.п.) - всегда активированные и дезактивированные ароматические соединения имеют свои методы, свои варианты реагентов и условий реакции.
А вот палладирование удивительным образом не так сильно различает между активированными и дезактивированными (в смысле обычного ароматического электрофильного замещения) субстратами. Совсем не различает? Нет, различает, но разница между анизолом и нитробензолом не более одного порядка константы скорости.
Данные по парциальным факторам скорости (если забыли, это относительные константы скорости в расчёте на одно положение,
приведённые к константе скорости на одно положение бензола) в рекции Фудзивары-Моритани, измеренные самими основоположниками через 10 лет после выхода первой статьи, очень хорошо показывают необычный характер этой реакции. Видим, что нитробензол дезактивирован (факторы меньше единицы), а анизол активирован (факторы кроме fm больше единицы), и в случае анизола видим орто/пара-ориентацию, а у нитробензола - мета. Всё вроде бы так, как в обычном электрофильном замещении, но, если приглядеться, как-то странно.
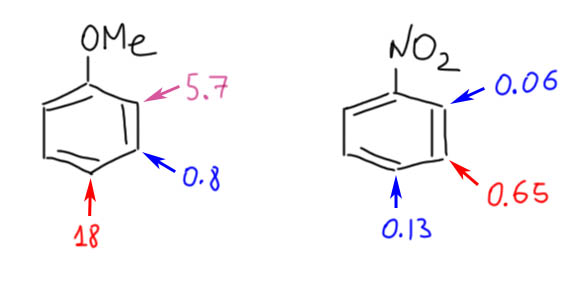
Во-первых, разница очень маленькая - между самым активным положением анизола и таким же нитробензола - всего два порядка. Это как раз и означает, что оба этих производных бензола можно вводить в реакцию в одних условиях с одним и тем же реагентом - для нитробензола нужно просто поднять температуру реакции на 20-30 градусов и подождать чуть подольше.
И парциальные факторы тоже сильно отличаются от обычных. В обоих случаях распределение изомеров намного более равномерное, чем в обычном электрофильном замещении - в смесях есть все возможные изомеры во вполне ощутимых количествах. Особенно бросается в глаза необычно большие факторы для "неправильных" положений, например, мета- для анизола. Судя по фактору скорости это положение практически не является дезактивированным, в то время как в обычном электрофильном замещении мета-положение при мезомерно-донорных заместителях всегда отличается на много порядков от орто/пара, и всегда сильно дезактивировано.
(на деле, мета-изомер в таких случаях образуется в следовых количествах, определяемых только высокоэффективной хроматографией). То же с нитробензолом. В обычной электрофильной химии "неправильные" изомеры тоже получаются (это даже является серьёзной проблемой для промышленности взрывчатых веществ - неправильные изомеры при нитровании, например, толуола приходится отделять, иначе получается некачественный и опасный продукт. Но не в таких количествах.
Странное распределение изомеров и небольшая разница в реакционной способности между активированными и дезактивированными производными бензола говорит о том, что у этой реакции особый механизм, и - открывает широкие возможности применения этой реакции и вообще реакций металлирования в синтезе
Итак, зависимость скоростей (парциальных факторов скорости) от заместителей показывает, что в основе реакция всё-таки имеет электрофильный характер (донор активирует, акцептор дезактивирует). Но слабая зависимость от заместителей, вполне очевидно, говорит о том, что палладий не лезет в ароматическое кольцо так, как это делают обычные органические электрофилы - сигма-комплекс (aka аренониевый ион, aka комплекс Уиланда) не образуется.
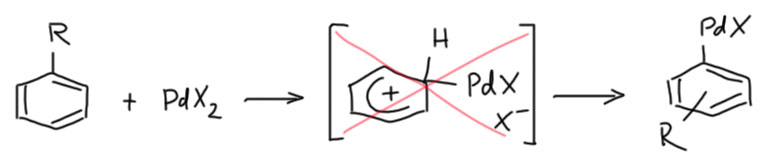
В принципе, это напоминает ситуацию с окислительным присоединением Pd(0) - тоже немного похоже на нуклеофильное замещение, но именно немного, слегка.
Реакция палладирования, как мы уже не раз видели в химии переходных металлов, также имеет согласованный характер. Поскольку нам нужно забрать у ароматического соединения протон, нам потребуется основание, а возможность отщепления протона обеспечит, как уже не раз бывало, электрофильная активация связи переходным металлом.
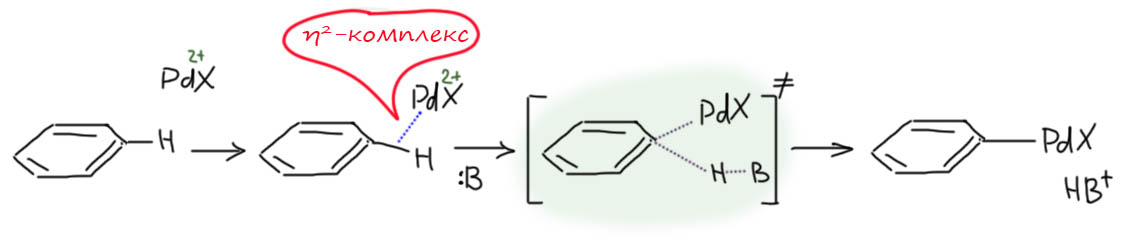
Можно предположить образование слабого комплекса с ароматической молекулой по C-H связи (некоторые исследователи предпочитают говорить о слабой, агостической связи металл-водород, но это схоластический спор, для нас точно не имеющий значения), и там как раз и произойдёт увеличение CH-кислотности. настолько, что протон отщепится слабым основанием, а палладий получит в своё распоряжение бывшую пару этой связи. Ароматическое кольцо, как видим, остаётся наблюдателем в этой реакции, а заместители на нём могут оказывать косвенное некоторое влияние на энергию интермедиатом и переходного состояния.
Простой согласованный механизм не даёт ответа на очень важный вопрос - где то основание, которое помогает удалить протон. Экспериментальные данные дают на это довольно недвусмысленный намёк - для палладирования годятся не все соли и комплексы палладия. Во всех известных на ранних этапах исследования примерах в реакционных смесях можно найти ацетат или что-то сильно на него похожее: либо сразу берут ацетат палладия, либо ацетат добавляют в реакционную смесь. Галогениды палладия совершенно бессильны, хотя с точки зрения электрофильности палладий в галогенидах вряд ли должен уступать палладию в ацетате.
Механизм, который это учитывает стал общепризнанным не только в палладировании, но и в металлировании комплексами других переходных металлов. Его принято называть CMD - Concerted Metalation-Deprotonation - Согласованное металлирование-депротонирование - СМД.
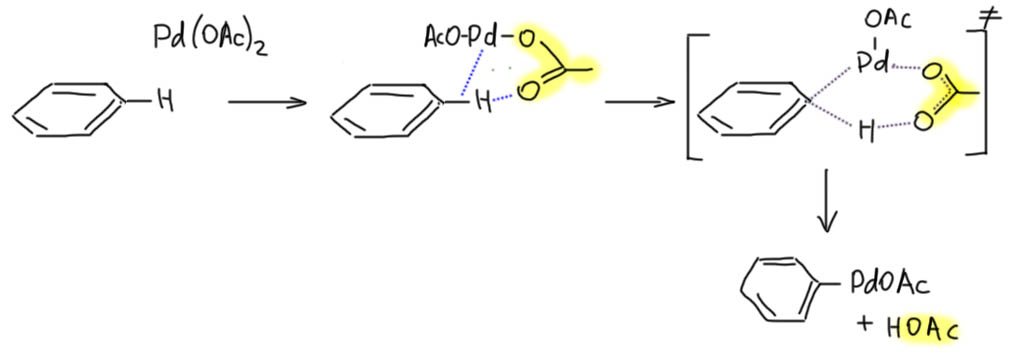
Смысл этого механизма в том, что ацетат, или что-то на него похожее, забирает протон в циклическом переходном состоянии, прямо из координационной сферы палладия. Все участники процесса - ароматическое соединение со связью C-H, палладий и ацетат - находятся в координационной сфере палладия. Сначала они туда входят за счёт координационных взаимодействий, и затем, по мере развития взаимодействий, ведущих к разрыву и образованию связей, дают продукт реакции.
В новом веке совершенно неожиданно началось возрождение интереса к реакции Фудзивары-Моритани. Были найдены системы, обеспечивающие регенерацию активного палладия(2+), часто использующие простые соединения меди(2+) или серебра(1+) как стехиометрические окислители и активаторы. Проблема низкой региоселективности решалась двумя способами. Первый, рассмотренный на этом слайде, просто взять субстраты, ограничивающие возможности образования изомеров положения. О втором, более мощном, поговорим на следующих слайдах.
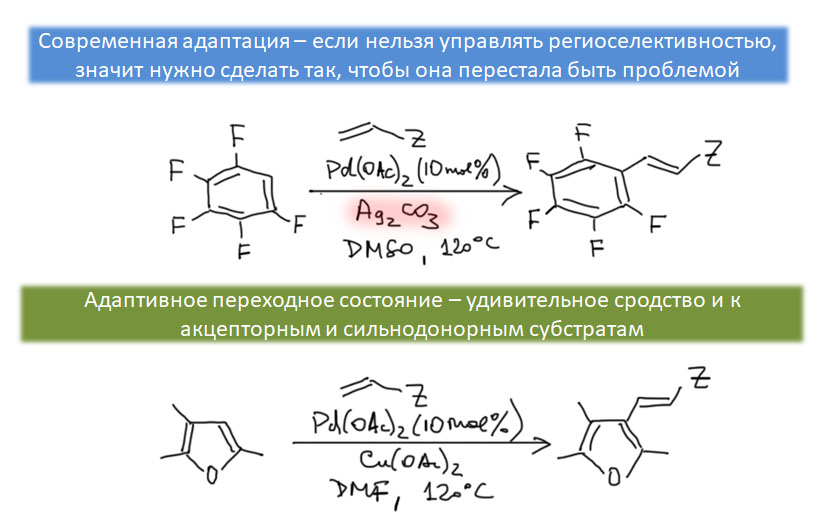
Поскольку новая, модифицированная реакция имеет общие черты с реакцией Хека, ее стали часто называть реакцией Фудзивары-Хека
У реакции в таком, новом обличье обнаружилось интересное свойство - она оказалась применима и к сильно-донорным, и к акцепторным ароматическим соединениям. В принципе, то, что палладирование применимо и к донорныи и к акцепторным субстратам, мы уже говорили. Но - есть одна интереснейшая особенность, которая говорит нам, что слабая зависимость от эффектов заместителей - это далеко не вся правда.
В современных протоколах реакции Фудзивары-Хека с почти равной вероятностью встречаются и донорные и акцепторные субстраты, и дело скорее не в том, что реакция почти одинаково относится и к тем, и к другим, а в том, что для каждого типа субстратов используются отдельные протоколы. Да, различие между ними не так велико, поэтому можно точно сказать, что речь идёт об одной реакции, а не о двух разных. Скорее, дело в том, что при небольших вариациях в протоколе мы можем получить то, что пригодно для каждого.
- каждому (субстрату) по потребностям!
Можно ли одним механизмом описать обе реакции обоих типов субстратов?
Ответ прост - конечно, да. Такое поведение не какой-то уникальный случай, оно нередко встречается в согласованных механизмах реакций и в самой обычной органической химии, и в химии переходных металлов. В последней оно особенно важно, потому что в химии переходных металлов почти все механизмы согласованные.
Такое поведение - мы как будто видим, что один и тот же реагент приспосабливается (адаптируется) к субстрату обычно принято описывать концепцией переменного переходного состояния. В обычной органической химии самый классический случай такого поведения - элиминирование E2. Мы знаем E1-подобный E2, E1cb-подобный E2, и просто E2 между этими крайними случаями - получается такой непрерывный спектр возможных структур переходного состояния. При желании, это можно даже красиво изобразить трёхмерными диаграммами, показывающими положение переходного состояния в зависимости от переменных, описывающих субстраты (это называется диаграммами Мо О'Феррала-Дженкса), но прока от таких диаграмм немного, так как параметры, используемые для их построения, чисто умозрительные. Поэтому можно обойтись без диаграмм, а просто словами описать вариацию структуры переходного состояния.
Один тип переходного состояния предполагает более тесное взаимодействие палладия с ароматическим кольцом - такой вариант механизма называют электрофильным CMD.
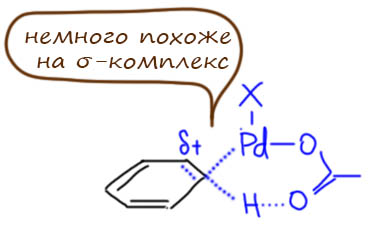
Этот тип механизма описывает реакции сильнодонорных субстратов, особенно всяких пятичленных гетероциклов. В производных бензола благоприятны мезомерные доноры. В целом, такое поведение можно считать образцово электрофильным, очень похожим на обычный механизм SEAr.
Этот тип механизма описывает реакции акцепторных субстратов, например, полифторзамещённых бензолов и акцепторных гетероциклов. Отщепляются обычно более кислые протоны, хотя на это очень большое влияние оказывает стерика.
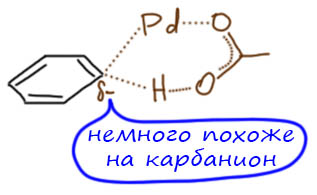
Другой тип переходного состояния предполагает более сильное взаимодействие лиганда-основания с протоном. Этот вариант механизма иногда называют стандартным CMD просто потому, что этот вариант делает акцент именно на депротонировании.
Но палладирование и другое металлирование переходными металлами имеет ещё одну особенность - наиболее легко оно происходит в направленном режиме. Эта реакция имеет вполне определённый прототип в химии непереходных металлов.
Гораздо более плодотворный подход использует направленное металлирование. Это явление очень хорошо известно в химии непереходных металлов, и весьма широко применяется в направленном литировании, методе получения литий-органических соединений прямым литированием ароматических соединений (в том числе гетероциклических), имеющих разные направляющие группы, в состав которых входят атомы с неподеленной парой.
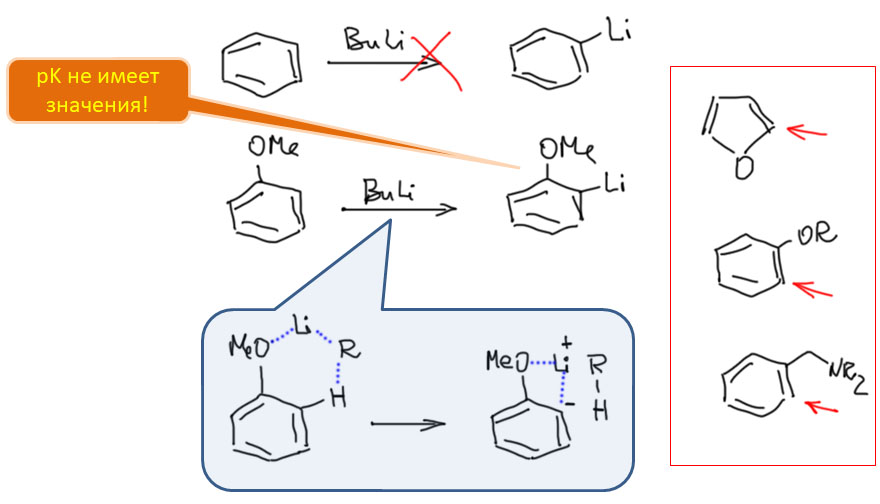
Характернейшей особенностью направленного металлирования является то, что оно имеет более высокий приоритет по отношению к другим факторам. Иными словами, когда идёт направленное металлирование, эффекты заместителей в остальной части молекулы имеют второстепенное значение и практически не учитываются.
Направленное палладирование - очень распространённая реакция. Дело здесь не в палладии - многие другие переходные металлы точно так же очень легко образуют металлациклы. Обратите внимание, что принято говорить и писать металлАцикл, а не металлОцикл, хотя второе более отвечает правилам русского языка, в котором нет других соединительных гласных кроме -о-. Но мы идём на поводу у английского языка, в котором, во-первых, таких ограничений нет, а, во-вторых, эта буква намекает на то, что металлацикл - это гетероцикл, а номенклатуре гетероциклов очень распространена заместительный принцип, и гласная -а- говорит о формальном замещении воображаемого атома углерода на гетероатом.
Палладациклы очень легко образуются при взаимодействии простых производных палладия с подходящими соединениями, в которых есть C-H связь для металлирования и направляющая группа на верёвочке подходящей длины. Металлируемая молекула фактически служит хелатным лигандом. Но хотя мы знаем, что в обычных хелатных циклах размер цикла практически может быть любым, в палладациклах чрезвычайно редко встречается что-то отличное от классических 5- и 6-членных циклов.
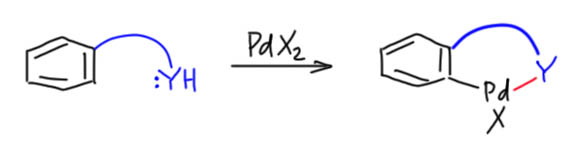
При этом ограничении разнообразие палладациклов совершенно ошеломительно. Эти соединения красивы и очень легко образуются, поэтому очень многим исследователям казалось, что именно в них Творцом всего сущего и Природой-матерью заложена какая-то фундаментальная тайна, добыть которую столь же прекрасно, как овладеть для средневекового рыцаря чашей Грааля. Наделали этих палладациклов несметное количество и очень долго пытались их пристроить к разным важным вещам, и очень часто получали совершенно банальные результаты. Только в новом столетии все немного успокоились и принялись за более практичные вещи, и на этом пути нарыли много интересного, хотя и не тянущего на нобелевские премии.
Напомню вам про один палладацикл, с которым мы уже встречались.
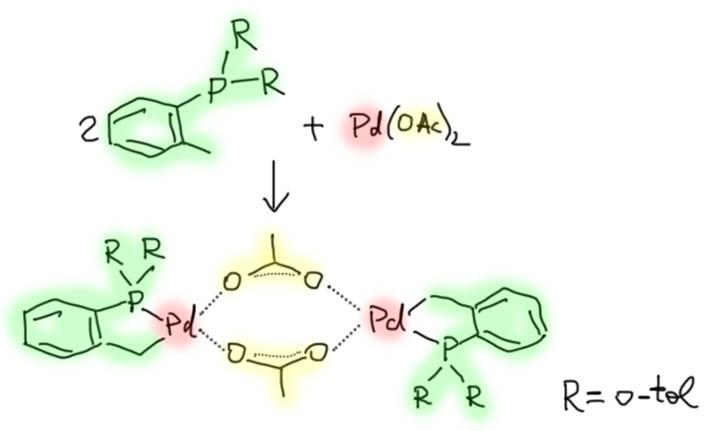
Палладацикл Херманна-Беллера, открытый, на самом деле Спенсером, очень легко получается из лиганда Спенсера, трис(о-толил)фосфина при взаимодействии с ацетатом палладия. Этот комплекс представляет собой димер (как и большинство других палладациклов) и обладает совершенно удивительной устойчивостью. Наверное, это самый знаменитый, и при этом один из самых бесполезных комплексов палладия. Бесчисленное количество химиков, по какой-то странной причине не умеющих считать электроны в комплексах, пали жертвой этого коварного комплекса, посчитав его well-defined катализатором нового типа. На деле, в реакциях он всегда разваливается, высвобождая бесфосфиновый палладий.
Палладациклы всегда образуются в реакциях с C-H активацией за счет направленного палладирования.
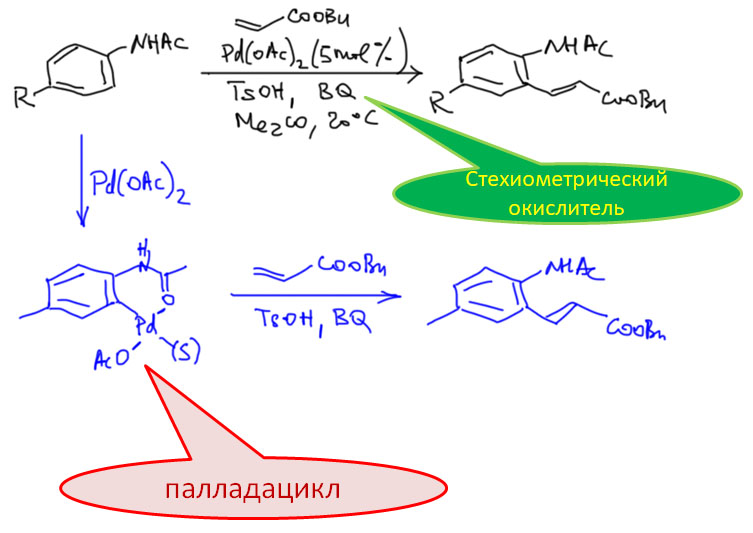
Присутствие направляющих заместителей в ароматической молекуле делает возможным проведение реакции Фудзивары-Хека в очень простых условиях, с использованием бензохинона в качестве стехиометрического окислителя. В качестве промежуточных продуктов в таких реакциях в результате внутримолекулярного направленного палладирования всегда образуются палладациклы. Реакцию всегда можно провести в две стадии - сначала получить палладацикл, и после ввести его в реакцию с олефином.
Комплексы Pd(2+) раньше других показали потенциал в прямом арилировании. Эти реакции известны с самых первых экспериментов по палладированию ароматических соединений. Эти реакции можно считать прямыми аналогами кросс-сочетания в том же смысле, в каком реакция Фудзивары-Моритани является аналогом реакции Мидзороки-Хека. Точно так же необходим дополнительный окислитель; нужны условия, способствующие проявлению электрофильности, и точно так же реакция малоселективная с простыми производными бензола, и вполне полезна, если место палладирования определено либо за счет заместителей, не оставляющих вариантов, либо за счет направленности палладирования. Очень популярной стала эта реакция в химии гетероциклических соединений. Реакцию чаще применяют в режиме гомосочетания, так как при реакции двух разных ароматических соединений получается еще и смесь гомо- и кросс-продуктов. Другое важное применение реакции - внутримолекулярное, когда кольца уже сближены, и палладию ничего не остаётся кроме как сшить их.
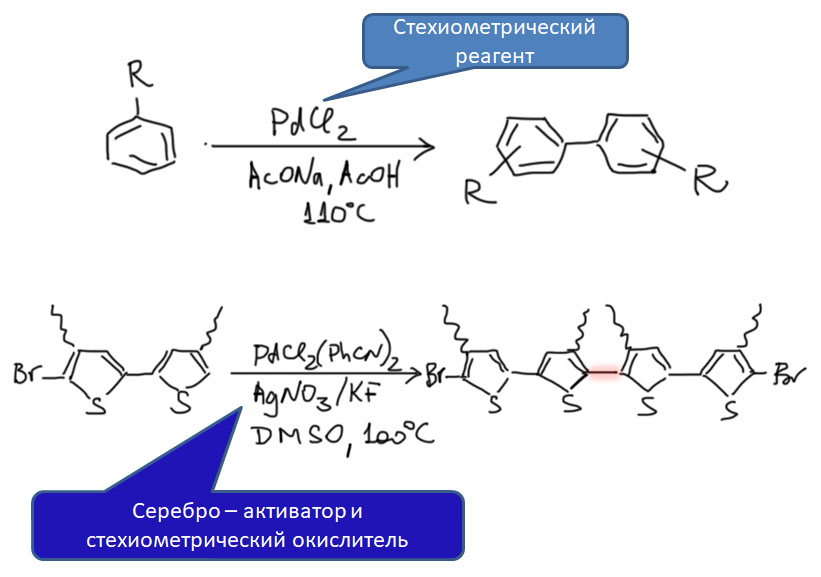
Прямое соединение двух ароматических соединений, каждого по C-H связям, безусловно, выглядит сбывшейся мечтой экономиста атомов, но реакция эта капризна, малоселективна по обоим субстратам, и вообще чудес не бывает, и экономисту придётся вернуться на место и продолжать мечтать. А мы пока займёмся чем-нибудь попрактичнее. и набьём, для начала, ещё пару шишек.
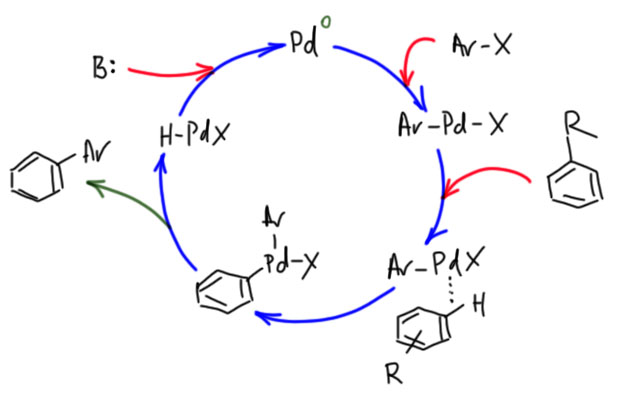
Давайте попробуем начать реакцию как обычное кросс-сочетание, окислительным присоединением Pd(0) к электрофилу. А вот дальше попробуем воспользоваться тем, что в аддукте Pd(2+) уже может обладать способностью к электрофильной атаке на C-H связь другого ароматического соединения. Более того, если мы всё это распишем, то поймём что нас ждёт что-то типа обобщения реакции Хека, и для замыкания цикла понадобится только основание. Очень похоже на Хека, не правда ли? Не совсем. В химии любые аналогии уместны, но всегда нужно знать когда остановиться. Ароматические соединения всё же не олефины, и у нас никуда не делась проблема селективности, да и в способность палладия в аддукте окислительного присоединения к атаке на CH есть некоторые сомнения.
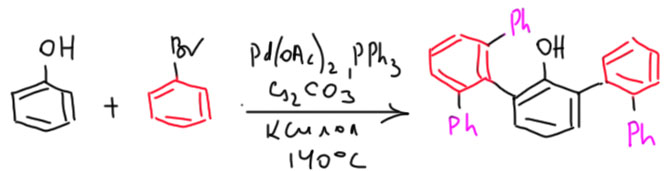
Первые попытки сделать эту реакцию появились ещё в проклятые девяностые, когда понимание сути процессов было еще невелико. Понятно, что брали ароматические соединения подонорнее. Уже знакомые нам Миура и Сато взяли фенолы. И да, обнаружили арилирование, причём совершенно очевидно направленное, потому что всегда получались орто-замещённые.
Kawamura, Y.; Satoh, T.; Miura, M.; Nomura, M. Chem. Lett. 1999, 961.
А если взять побольше бромбензола то первоначальный продукт направленно арилировался дальше с образованием такой развесистой молекулы. И почему туда не вошёл ещё один фенил, понять трудно, но видимо в такой тесноте уже не помещался ещё один комплекс палладия.
В целом реакция до начала нового столетия оставалась курьёзом без серьёзных перспектив.
Межмолекулярное палладий-катализируемое арилирование в направленном варианте почти всегда страдает от образования смесей продуктов моно- и диарилирования, если только второе положение не заблокировано. Поэтому его обычно применяют тогда, когда нужен именно продукт диарилирования, так как этого можно добиться использованием избытка электрофила. Иногда, впрочем, доступных мест может быть и больше.
Самый крутой фокус этого типа показал Хартвиг. Ему в какой-то момент стало обидно - его вечный конкурент Бачуолд наплодил целое семейство модных лигандов с красивыми названиями - JohnPhos, DavePhos, BrettPhos, RuPhos и т.п. А достижения Хартвига были не менее велики, но вот модных лигандов долго не было. И тогда Хартвиг взял и проарилировал дитретбутилфосфиноферроцен.
Хлорбензол стал и растворителем и реагентом, а само исходное и лигандом и направляющей. Донорный лиганд помогает палладию окислительно присоединиться к хлорпроизводному. Ну и дальше просто представьте, как верхний блин с ручкой, на которую прицепился палладий, объезжает по кругу нижний блин, скидывая ему фенил, тут же хватая новый, снова скидывая, и так далее, сколько влезет. Пожалуй, это самая комическая реакция из всех, которые я когда-либо видел. Но лиганд получился отменный, только фантазии у Хартвига хватило лишь на Q-phos.
Но действительно полезной и популярной реакция арилирования стала во внутримолекулярном варианте, когда промежуточный палладацикл образует новый цикл. Систематическое исследование таких реакций начал канадский исследователь Кейт Фанью, и он же внёс основной вклад в продвижение этой химии в синтез. В первых работах использовались специфические лиганды типа 2'-диметиламино-2-дифенилфосфинодифенила, но в дальнейшем стали применять более стандартные лиганды.
Campeau, L.-C.; Parisien, M.; Fagnou, K. J. Am. Chem. Soc. 2004,126, 9186
Наверное, самая яркая реакция, а точнее, целое семейство каскадов, были открыты и изучены итальянской исследовательницей Мартой Кателлани из знаменитого города Парма. Каскады Кателлани используют один приём - если ввести в реакцию Мидзороки-Хека норборнен, то из-за невозможности β-гидридного элиминирования аддукт карбапалладирования зависает, и ему ничего не остаётся, кроме как в бессильной злобе укусить то самое бензольное кольцо, которое его собственно туда и притащило. Образуется палладацикл, причем очень забавный - с двумя углеродными атомами, соединёнными с палладием. Это тоже трагедия для палладия - он должен сбросить это ярмо в восстановительном элиминировании, как он всегда делает с двумя углеродными лигандами. Но - не может, геометрия комплекса не даёт. Тогда бессильная злоба превращается в ярость, подкреплённую весьма немаленькой донорностью двух углеродных лигандов. И Pd(2+) становится способным к ... окислительному присоединению, правда, не арилгалогенида, а высокореакционноспособного алкилгалогенида. Образуется долгожданный комплекс Pd(4+).
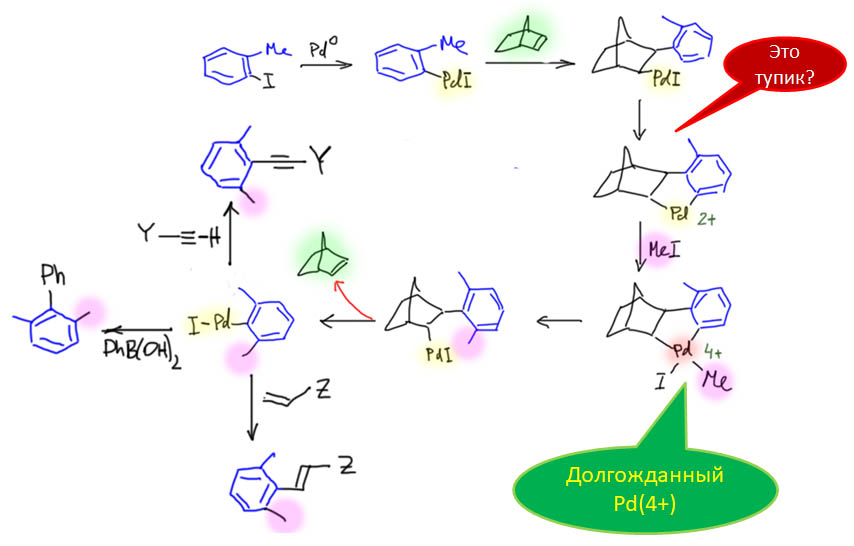
Три углеродных лиганда на металле с такой высокой степенью окисления - это точно перебор, и дальше происходит востановительное элиминирование, палладий опять возвращается на каркасный остаток норборнана, с которого нет гидридного элиминирования. Этому интермедиату уже больше некого кусать и он горестно разваливается на исходный норборнен и новый арилпалладий (да, карбопалладирование тоже обратимо), а его уже можно подловить на обычного Хека, на Соногасиру, на Судзуки-Мияуру, на карбонилирование, или чёрт знает на что ещё.
И что мы получили в результате такого головолокружительного мельтешения палладациклов? В общем, ерунду - почти то же самое, что и без него, только в орто-положениях появятся простые алкилы. Современная химия не станет так мучиться, потому что можно прямо сделать нужную реакцию из орто-замещенных субстратов. Но в 1990-х еще не было лигандов, которые позволяли вводить в реакцию стерически затруднённые субстраты. Сейчас есть. Так делать никто больше не будет, но изящество мысли и изощрённость каскада не могут не вызвать восхищения к работам Кателлани. Это такое чистое искусство, и недаром она работает и живёт в Парме, городе наполненном великолепной живописью и архитектурой.
В современном синтезе добиваются очень высокой реакционной способности каталитических систем, в частности, используя не случайные пути предактивации в смесях простых солей или комплексов палладия с фосфинами, а пред-катализаторы, которые в условиях реакции быстро генерируют высокоактивный монофосфиновый (или моно-NHC) катализатор. С одним типом таких предкатализаторов мы познакомились в аллильном замещении. Бухвальд предложил другой тип, основанный на палладациклах, с использованием восстановительного элиминирования как основного пути предактивации.
Бухвальд с сотрудниками в 2008-2013 годах последовательно предложил три поколения таких предкатализаторов. Каждое последующее было стабильнее, эффективнее, гибче, но в практике катализа в органическом синтезе прижились и заслужили немаленькую популярность все три. Их применяют в реакциях кросс-сочетания всех типов особенно тогда, когда нужны побольше TON и TOF, помягче условия, когда попадаются малореакционноспособные или не очень стабильные субстраты. Чаще всего предкатализаторы этого типа используют вместе с фосфинами Бухвальда (JohnPhos, XPhos, SPhos, BrettPhos, JackiePhos и т.п.), но никто не мешает с их помощью вводить и другие монодентатные фосфины и NHC-карбены.
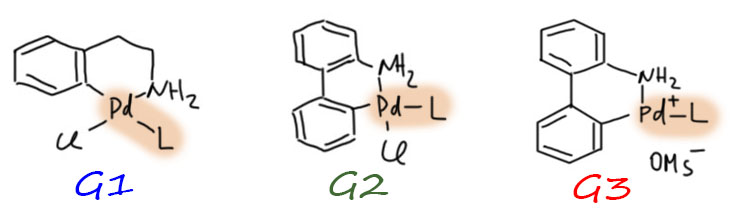
Пинцерные металлациклы.
Не менее интересны и такие представители палладациклов, и даже шире, металлациклов, в которых не одна, а сразу две направляющие группы. Такие соединения называются пинцерными металлациклами, и хотя и не сразу, но в последние лет 15 эти соединения получили большую известность, так как с ними связаны совершенно необычные каталитические и стехиометрические реакции в синтезе. Жесткая структура пинцерных металлациклов не дает им участвовать в реакциях кросс-сочетания, Мидзороки-Хека и т.п. без разрушения, но может быть очень полезной в других реакциях, где остающихся координационных возможностей достаточно, а жесткий контроль координационной сферы ограничивает число возможных путей превращения. За последние 15 лет обнаружено множество реакций, критически зависящих именно от пинцерной структуры катализаторов.
Направленное металлирование становится совсем однозначным, когда направляющие группы расположены с обеих сторон. В этих случаях реакции металлирования идут совсем легко, а образующиеся комплексы обычно обладают совершенно невероятной устойчивостью во всех смыслах этого слова, включая кухонно-бытовой (можно достать вещество из колбы и хранить без всяких предосторожностей, хоть в солонке или перечнице), независимо от того, каким был лиганд (например, пирофорный и токсичный триалкилфосфин) и металл (в таких комплексах бывают металлы в необычных степенях окисления).
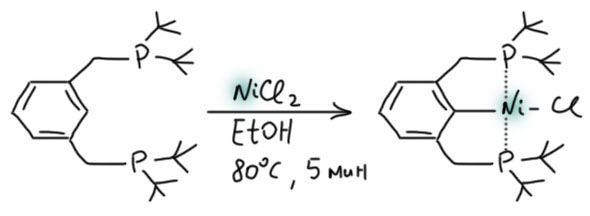
C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020
Впервые несколько комплексов этого типа получил английский исследователь Бернард Шоу (не нобелевский лауреат и не драматург). Условия образования были настолько просты и мягки, а вид новых комплексов настолько красив, что Шоу пришёл в неописуемый восторг и решил, что он открыл нечто совершенно выдающееся, то, что перевернёт всю химию. Восторг оказался заразен, и это привело впоследствии к большому количеству необдуманных выводов и несвершившихся открытий.
Ещё более серьёзный импульс исследованиям комплексов этого типа дал весьма влиятельный голландский исследователь Герард ван Котен, который показал, что металлабициклы этого типа можно получить не только из фосфинов, но из намного более простых исходных, например, аминов. Работы ван Котена очень ясно показали, что это действительно весьма общий структурный тип.
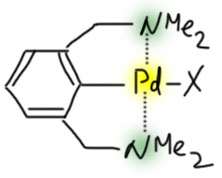
G. van Koten, et al. J. Chem. Soc., Chem. Commun., 1978, 250

Ван Котен же и придумал название пинцерные, то есть похожие на клещи. Есть подозрение, что этот выдающийся учёный никогда не держал в руках настоящих клещей, иначе бы ему не могло не прийти в голову, что самая главная черта новых комплексов - центральная связь - не имеет никакого воплощения в метафоре клещей, а сама по себе метафора клещей ничего не добавляет к метафоре хелатный (по-русски совсем забавно: клешня - клещи, - что в лоб, что по лбу). Но так или иначе, термин прижился.
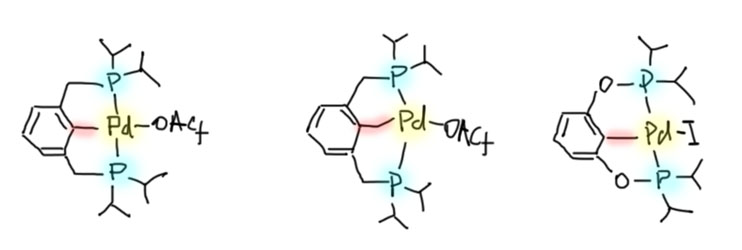
Пинцерными комплексами стали называть сначала любые металлобициклы, совершенно неважно от какой структуре произведённые и первоначальный образ таких лигандов как ароматического кольца с двумя ручками, обхватывающими металл, быстро ушло. Обратите внимание, что хотя иногда и лиганды называют пинцерными, это проявляется только в металлабициклических комплексах (двойных хелатах). Тот же лиганд в другом комплексе отлично может быть или простым хелатором, или мостиком, или чем угодно ещё. Пинцер - это всё-таки сам комплекс, а не его лиганд. Вот несколько примеров.
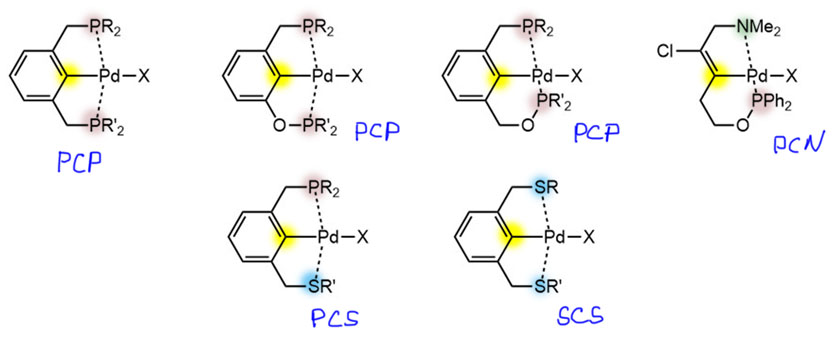
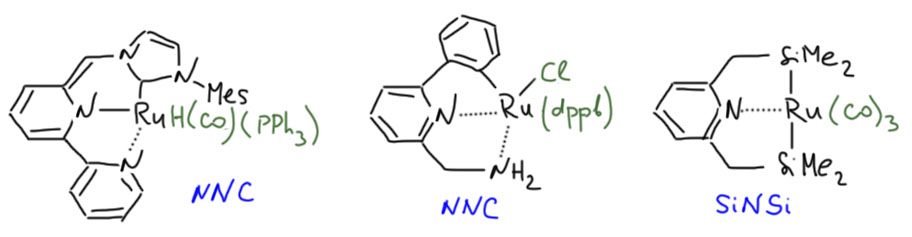
Пинцерные комплексы для удобства часто символически обозначают тремя атомами, обеспечивающими связь с металлом - центральный должен быть в середине. В начале в середине всегда был углерод, и комплексы образовывались всегда в результате циклопалладирования. Затем соблазн называть так любые комплексы с тридентатными лигандами и конденсированными хелатными кольцами победил, и пинцерами стали называть любые такие комплексы вообще. Вот ещё несколько примеров.
Когда пинцерные комплексы появились, желание запихнуть куда-нибудь такие красивые соединения стало просто навязчивой идеей для многих амбициознах исследователей. Одним из первых стал израильский химик Давид Мильштейн, тот самый, который был соавтором самого Стилле в великой статье, описавшей кросс-сочетание с оловоорганикой. Мильштейн заявил, что один такой пинцер, не самый простой, содержащий высокодонорные триалкилфосфиновые центры, является просто невероятным катализатором реакции Мидзороки-Хека (D. Milstein et al, J. Am. Chem. Soc., 1997,119, 11687). Эта статья стала одной из важнейших в той "гонке за TON", которую мы уже обсуждали.
Единственное, что оставалось не ясно, это вопрос, а как такой комплекс может катализировать реакцию Хека, для которой, как мы уже выяснили, нужны три доступных места, в то время как Мильштейн и его последователи настаивали на том, что пинцерные катализаторы имеют абсолютно устойчивую структуру, то есть доступно только одно место. Уже упоминавшийся Бернард Шоу (не драматург) пытался выдвинуть концепцию нового механизма реакции Хека через Pd(4+) и Pd(2+), но доказать этого не смог ни он сам, ни его последователи.
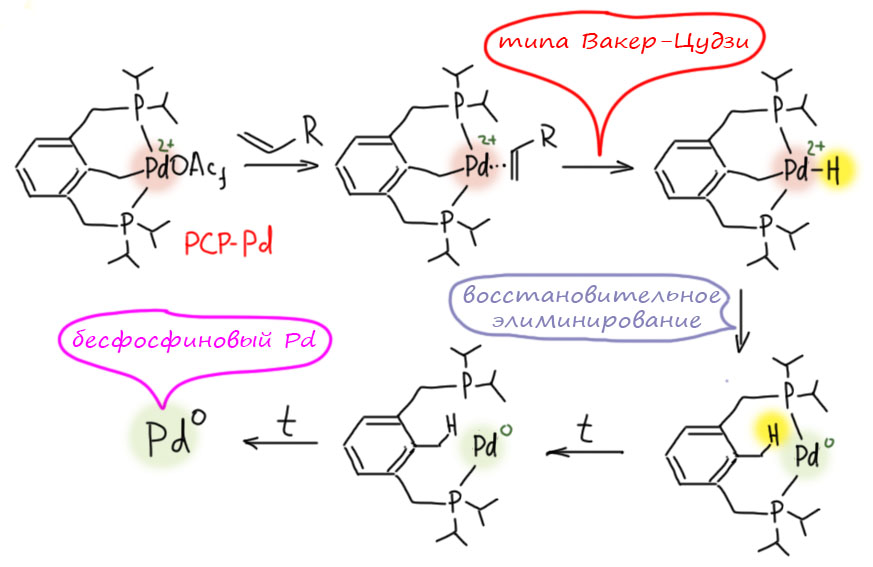
В 2000 г И.П.Белецкая и автор этого курса, проанализировав все данные по катализатору Мильштейна и уже тогда ставшими многочисленными похожими системами предположили, что эти структуры не так стабильны, как хочется их создателям, и что на самом деле все эти системы работают как ... бесфосфиновые, резервирующие активный палладий в виде наночастиц.
Разборку комплекса Мильштейна в условиях реакции Хека можно представить, например, так. Начинается всё с реакции с олефином (реакции Вакер-Цудзи), причём олефин окисляется, а палладий получает гидридный лиганд. Восстановительное элиминирование фактически обращает тот процесс, который и привёл к образованию пинцера. А при высокой температуре реакции оставшиеся фосфины очень быстро просто отвалятся и превратятся в фосфиноксиды.
Восстановительное элиминирование фактически обращает тот процесс, который и привёл к образованию пинцера. А при высокой температуре реакции оставшиеся фосфины очень быстро просто отвалятся и превратятся в фосфиноксиды. А это, в свою очередь, значит, что модный пинцер и тьма его последователей даром никому не нужны, потому что бесфосфиновый палладиевый катализ можно запустить намного проще и дешевле.
Эта идея сначала была встречена с возмущением, гонка за TON и производство "новых катализаторов" продолжались еще несколько лет, но стала понемногу выдыхаться, и к концу нулевых концепция бесфосфинового катализа стала общепринятой. А такие мощные учёные как Мильштейн бросили эту забаву сразу и занялись действительно интересной химией.
Когда серьёзные исследователи, в первых рядах сам Мильштейн, осознали, что простых путей куда-то пристроить пинцеры нет, они занялись глубоким изучением их свойств и каталитической активности. Стало ясно, что нужно целенаправленно искать процессы, в которых жёсткая координационная структура пинцеров станет настоящим преимуществом. В последние 15 лет дело для пинцеров нашлось и не одно.
Очень активно стали развиваться реакции дегидрирования, для которых пинцерные комплексы рутения и иридия дают просто потрясающие достижения. Реакция дегидрирования превращает, например, алкан в алкен, или спирт в альдегид или кетон. Хороших катализаторов дегидрирования, способных работать в мягких условиях, известно было немного. Исследования Мильштейна по пинцерным катализаторам внесли огромный вклад в эту область. И не только результаты, но и сами катализаторы производят большое впечатление.
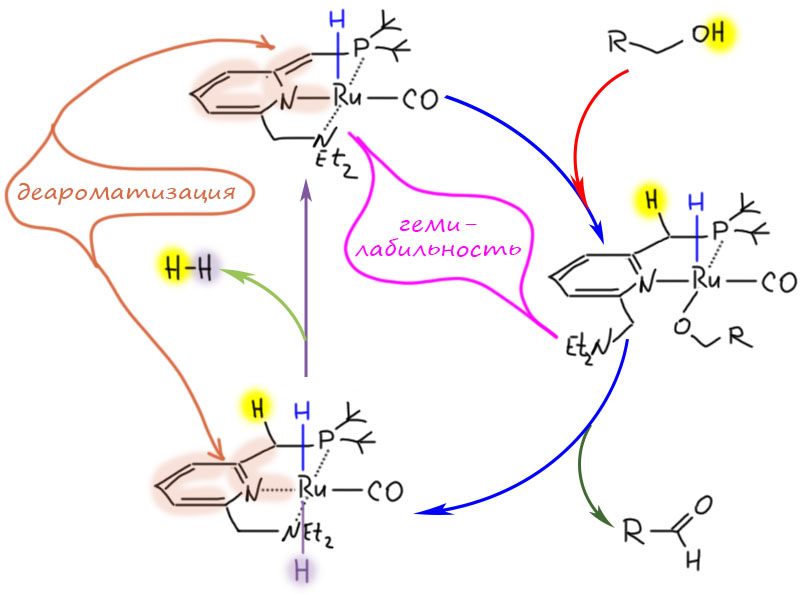
Zhang, J.; Gandelman, M.; Shimon, L. J. W.; Rozenberg, H.; Milstein, D. Organometallics 2004, 23, 4026
Вот, например, NNP-пинцерный комплекс рутения, творящего чудеса в реакциях дегидрирования. Со спиртами, например, он реагирует, отнимая у них водород в два захода, причём в деле участвует не только центральный атом металла, но и сам лиганд, по ходу дела превращающийся их производного ароматического пиридина в неароматическое производное дигидропиридина. Такое процесс в современной химии стало модно называть деароматизацией - намёк на то, что в деле участвует процесс, который раньше было принято считать невыгодным и всегда идущим в обратном направлении - в сторону ароматизации. Именно лиганд забирает первый атом водорода со спирта.
Такое поведение лиганда один из самых знаменитых основоположников металлоорганической химии переходных металлов Роберт Крэбтри, предложил игриво называть non-innocence, то есть буквально не-невинность, что лучше трактовать через уголовно-прецессуальное словоупотребление, чем какое-то другое.
Кто украл водород? - обращаемся мы к катализатору, ожидая, что как обычно это сделал металл. Но нет, есть у металла водород, да не тот, хотите пометьте, если не верите. И тогда наши взоры обращаются к лиганду - мы не верим в его виновность, потому что знаем из всей предшествующей химии, что анциллярные лиганды сами ничего не делают, кроме того, что в уголовном праве называется пособничеством. Но кто-то же слямзил водород! А, вот он водород, да и сама природа лиганда изменилась именно для того, чтобы дать возможность рутению принять алкоксильный лиганд. И вот ещё одна особенность мильштейнова пинцера - он, Мильштейн, очень хорошо усвоил урок, что пинцеры не такие железобетонные, как он утверждал в начале своей блестящей карьеры. Пинцер может очень хорошо приспосабливаться, и становиться гемилабильным, убирая самую слабую свою руку от металла, чтобы дать место более важному лиганду-актору. Дальше жадный до водорода рутений бесцеремонно выдирает его у алкоксида по хорошо нам знакомому механизму бета-гидридного элиминирования, отпуская альдегид. Цикл может замкнуться выбросом диводорода и деароматизацией. Для этого нужны некоторые дополнительные усилия, которые мы пока опустим. Этот небольшой цикл и так уже производит неизгладимое впечатление - это молекулярная эквилибристика высшей пробы!
В реакциях дегидрирования водород обычно не выделяется в виде газа, а уходит на какой-нибудь акцептор - алкен, альдегид, ещё что-нибудь. В лаборатории это нормально - такие реакции, их называют transfer hydrogenation/dehydrogenation (по-русски это можно перевести или длинно или коряво,) - очень любят, потому что в лаборатории не любят связываться с газом водородом. А вот для масштабных применений лучше как раз, чтобы выделялся именно водород, а не какая-то непонятная дрянь, которую ещё нужно утилизовать.
Но есть и ещё более невероятное применение реакций дегидрирования - в качестве аккумулятора и источника водорода. Водород - идеальное топливо для двигателей, но его очень трудно и опасно транспортировать, и многочисленные способы его запасать в сорбентах различной природы пока не привели к надёжному решению.
Поэтому на полном серьёзе исследуются реакции гидрирования/дегидрирования удобных соединений: гидрирование запасает водород, дегидрирование высвобождает его. И выяснилось, что такие системы можно сделать практичными если а) у нас есть эффективная каталитическая система для обратимого гидрирования/дегидрирования; б) субстратом для этой системы является простое и обязательно жидкое органическое соединение. Такие соединения прозвали жидкими органическими резервуарами водорода (liquid organic hydrogen-storage compounds, LOHC). И очень интенсивно исследуют. Попутно часто получаются полезные и необычные реакции.
Вот, например, как превратить спирт в сложный эфир. Катализатором является тот же рутениевый пинцер Мильштейна.
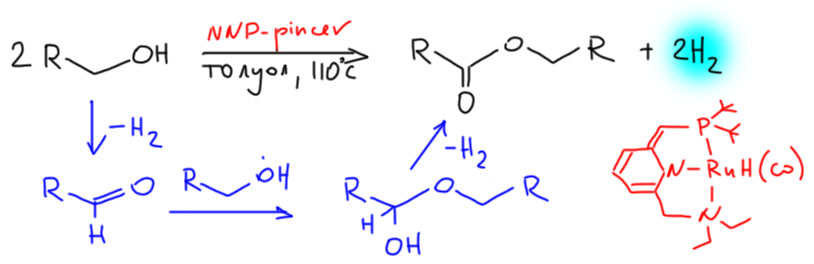
Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. J. Am. Chem. Soc. 2005, 127, 10840
Реакция имеет неплохую эффективность - TON до 1000. Сначала спирт дегидрируется в альдегид, который даёт со спиртом полуацеталь. В обычной органической химии мы сталкивались с полуацеталями, которые спонтанно образуются в равновесии, но неустойчивы и никаких применений поэтому не имеют. А вот рутениевый пинцер отлично может перехватить полуацеталь и, дегидрировав его, превратить в сложный эфир. Таким образом это типичный тандемный процесс: первый каталитический цикл превращает спирт в альдегид каталитическим дегидрированием, дальше альдегид в обычной равновесной стехиометрической реакции даёт полуацеталь, и последний вступает в другой каталитический цикл дегидрирования с образованием сложного эфира.
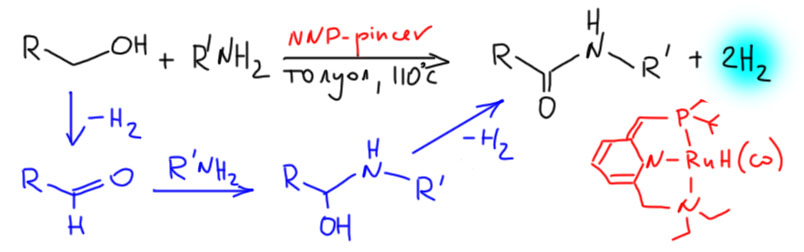
Ещё интереснее применение пинцерного катализатора для прямого получения амидов из спиртов и аминов. Водород отщепляется со стороны спирта. Реакция тоже идёт через альдегид и полуаминаль за два захода, то есть фактически является тандемом: дегидрирование - полуаминаль - дегидрирование.
Этот метод успешно применили к бета-аминоспиртам и получили или пептиды, или известные из химии аминокислот дикетопиперазины - циклические дипептиды. Из самого простого аминоэтанола получается дикетопиперазин глицина, и эта молекула действительно неплохо соотвествует искомым кандидатам на LOHC - она маленькая, и на неё приходится целых 4 молекулы водорода - в 114 граммах запасается почти сто литров газообразного водорода.
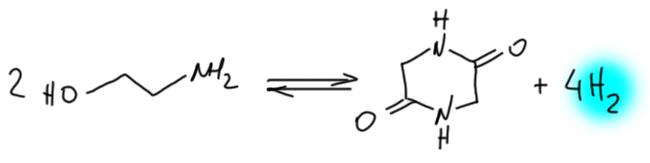
D. Milstein et al. Nature Commun. 2015, 6, 6859
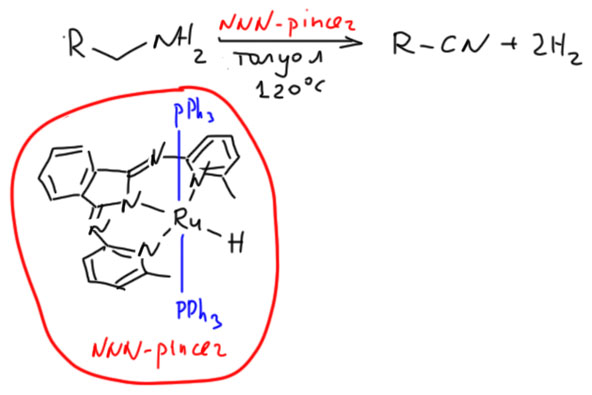
Tseng, K.-N. T.; Rizzi, A. M.; Szymczak, N. K. J. Am. Chem.Soc. 2013, 135, 16352
Ещё более невероятную реакцию дегидрирования описал другой весьма активный в этой области исследователь из США Натаниэль Шымчак (в польских фамилиях жиши можно и даже нужно писать с буквой ы). Рутениевый NNN-пинцер, производный от весьма популярной системы фталодиимида, просто так берёт - и отдирает два водорода от аминов с первичной алкильной группой, образуя ... нитрилы и две молекулы водорода. Реакция получилась вполне общая и имеющая неплохие перспективы в органическом синтезе. Мы обычно делаем наоборот - нитрилы гидрируем в амиды, и для этого есть тысяча и один способ. А вот наоборот смог впервые сделать только Шымчак, хотя по его следам фокус смогли повторить уже несколько других групп, и реакция получила неплохое развитие.
Мы уже поняли, что ничего практичного из углекислого газа не сделать. Но мучения с этой упёртой молекулой приводят к интересным решениям, имеющим самостоятельное значение.
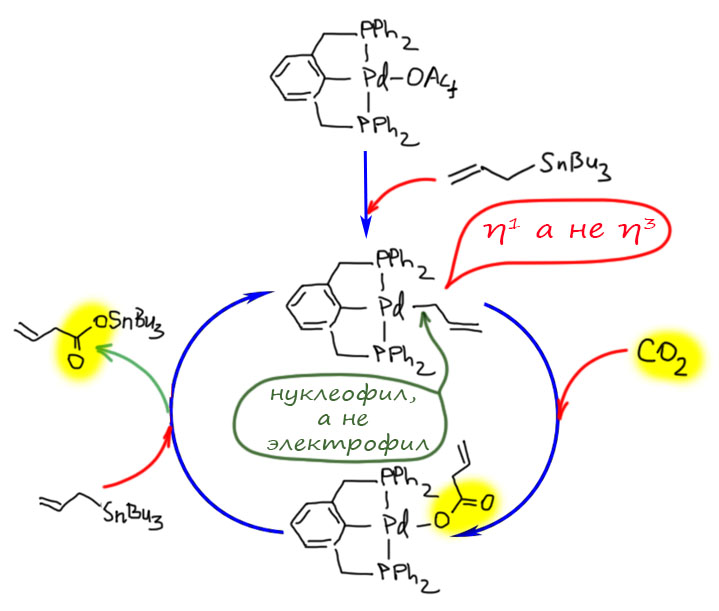
R. Johansson and O. F. Wendt, Organometallics, 2007, 26,
2426
Шведские исследователи Вендт и Йоханссон, использовав жёсткий палладиевый пинцер, заставили аллил закрепиться в η1-конфигурации, так как в этом случае обычная η3-конфигурация невозможна. И такой аллил становится тем, чем и должен быть σ-связанный органический лиганд - нуклеофилом, скрытым карбанионом. И он реагирует с углекислым газом как какой-нибудь гриньяр. И реакция даже может продолжаться в каталитическом цикле, так как аллилолово переметаллирует и снимает продукт реакции с палладия. Получается оловянная соль карбоновой кислоты, которую можно подкислить и получить саму карбоновую кислоту. Видите какое невероятное достижение в связывании углекислого газа. Так и видишь эту реакцию в масштабе пары десятков миллионов тонн хотя бы. И целое море оловоорганики в нагрузку. Выживет ли хоть какое живое существо на планете после такой утилизации парникового газа?
Но реакция интересная и показательная. За это и ценим.
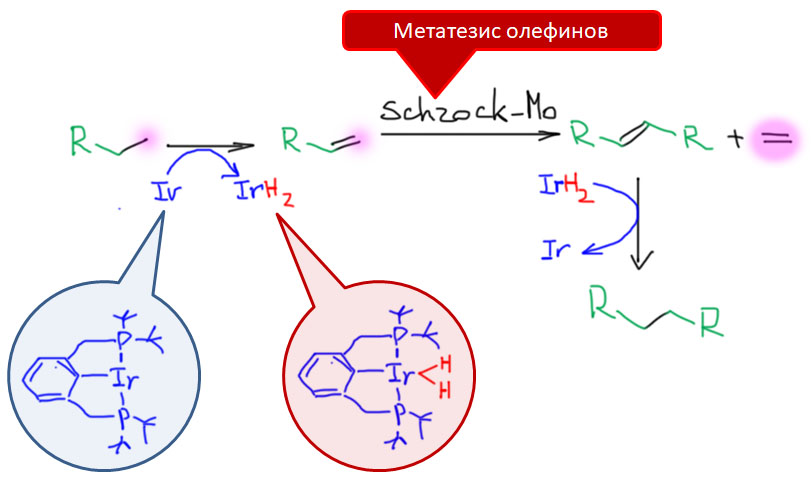
И на закуску - один из самых впечатляющих трюков, выполненных пинцерными каталитическими системами.
Один из примеров процесса, в котором пинцерный металлацикл играет ключевую роль, это метатезис алканов за счет трех последовательных реакций (тандемный процесс) дегидрирования, метатезиса олефинов и гидрирования. За первую и последнюю стадии отвечает иридиевый PCP-пинцер, который за счет фиксированной структуры и значительного стерического объема окружения металла селективно осуществляет дегидрирование концевой этильной группы, но гидрирования внутренней двойной связи.

Палладий велик, спорить с этим будет только невежда, но в славном деле CH-активации в новом столетии у него появился опаснейший конкурент. Палладий проспал этот наезд. Он всегда боялся конкуренции со стороны соседа справа – родия. И не без оснований. Родий давно обогнал палладий во всяких промышленных реакциях, и на этом сделался самым дорогим металлом в мире (я имею в виду полезные металлы, а не всякую экзотическую шелупонь, которая дорого стоит только потому что никому не нужна – родий, как и другие благородные металлы торгуется на самой настоящей бирже, и серьёзные люди вкладывают в него последние миллионы). Но в тонком органическом синтезе выскочил вперёд самый скромный из платиновых металлов – рутений. CH-активация на комплексах рутения – одна из самых модных тем в последние 10 лет. Не проходите мимо. Это отчасти похоже на палладий, но здесь всё по-другому, а возможностей не меньше, а больше.
К концу прошлого века вся химия, в которой образуются новые связи C-C, то есть именно то, что нужно органическому синтезу, прочно закрепилась за палладием, в тени которого иногда мельтешил его бедный родственник никель. Кросс-сочетание, Хек, карбонилирование, аллильное замещение, каскады - всё это обслуживал палладий, все говорили только про палладий, и сама область "комплексы переходных металлов в органической химии" фактически была химией палладия.
В 1990-е стали появляться отдельные работы, показывавшие, что и другие металлы могут делать что-то подобное. Но работы эти обычно проходили почти незамеченными и быстрого продолжения не имели. Эти работы всегда были очень узкими, и даже если находили какой-то новый каталитический процесс, он был сильно ограничен специфическим типом субстратов.
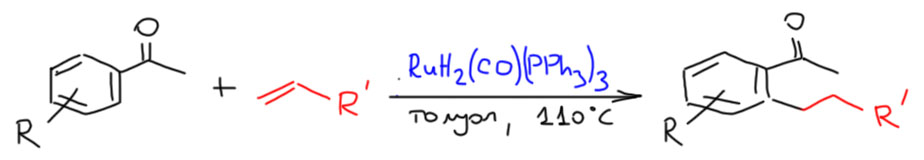
В 1993 году в самом гламурном научном журнале Nature появляется совместная работа нескольких японских групп под руководством Синдзи Мураи, Фумитоси Какиути и Наото Хатани (Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Nature 1993, 366, 529-). В этой реакции ароматический кетон реагировал с алкеном в присутствии рутениевого катализатора - результат был очень похож на реакцию Фудзивары-Хека, но с гидрированием двойной связи.
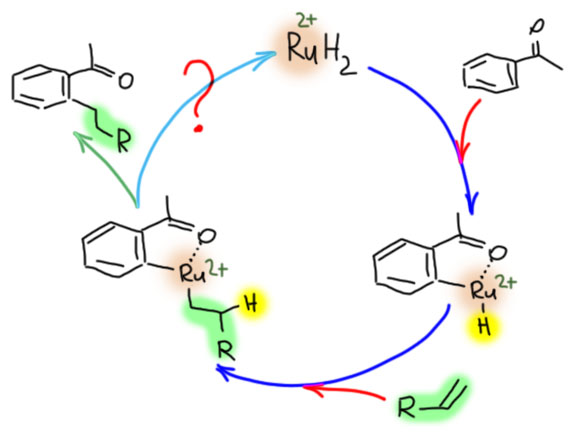
Сходство это обманчиво - реакция Мураи-Какиути-Хатани имеет совсем другой механизм, и C-C связь в ней образуется не за счёт бета-гидридного элиминирования, а за счёт восстановительного элиминирования. Начинается реакция, впрочем, точно так же - с направленного металлирования. Затем происходит миграционное внедрение - гидрорутенирование двойной связи. Ранним исследованиям в этой области (оцените заодно скорость развития - это было меньше 30 лет назад, но мы считаем это "ранним исследованием") свойственно плохое понимание превращений координационной сферы, поэтому в цикле не обозначены анциллярные лиганды. И совсем непонятно, как замыкается цикл и откуда рутений опять берёт гидрид. Реакция вполне каталитическая, использует 1-2 моль% пред-катализатора, но как замыкается цикл не ясно.
Плохое понимание механизма каталитичекого цикла не позволило быстро развить этот впечатляющий успех. Пришлось, как всегда в этой области, подождать, пока это понимание появится.
Но аналогии с палладиевым катализом оказались неудачными. Больше 10 лет понадобилось на то, чтобы разобраться как действует рутений в этой реакции. Интуиция не подвела - если в каталитическом цикле что-то потеряно и он не сходится, то это неверный каталитический цикл. Возможности рутения оказались - как бы крамольно это не прозвучало для тех, кто еще в прошлом веке привык к мысли о том, что круче палладия в этой химии нет и не может быть ничего - намного интереснее и даже парадоксальнее. Загадка потерянного гидрида разрешилась очень просто.
C-H активация происходит не за счет электрофильного металлирования Ru(2+), а за счёт нуклеофильного окислительного присоединения Ru(0) к связи C-H. Тоже направленного.
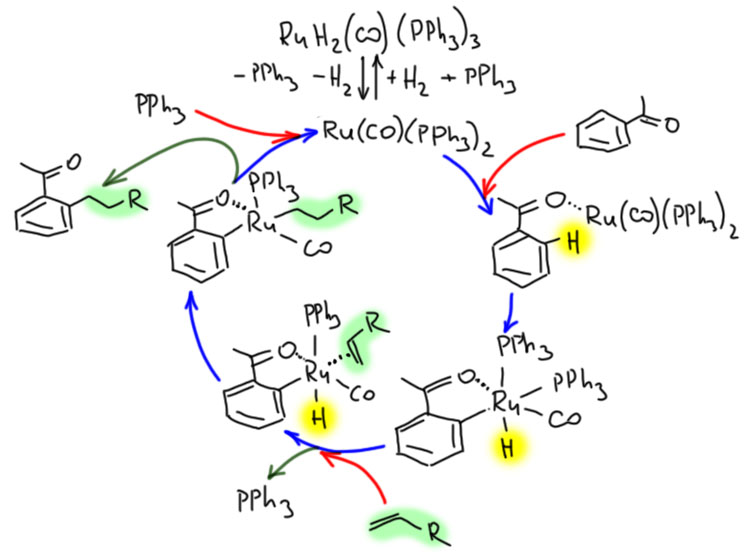
И на стадии предактивации пред-катализатор, комплекс Ru(2+) просто равновесно диссоциирует на водород и комплекс Ru(0), который и является истинным катализатором. Интересно, что в комплексе Ru(0) сохраняет способность координироваться по кислороду ацильной группы. Можно предположить, что эту способность поддерживает карбонильный лиганд, частично снимающий электронную плотность с Ru(0), создавая на металле кислотность Льюиса, обычно несвойственную низковалентным состояниям металлов.
Ru(0) окислительно присоединяется к связи C-H. Ничего невозможного в этом нет , хотя это и удивительно. C-H связи редко являются реакционным центрами для нуклеофильной атаки. Но в данном случае этому способствует пространственная сближенность. Дальше происходит ожидаемое связывание олефина и миграционное внедрение, за которым следует восстановительное элиминирование. Эта часть каталитического цикла оказалась вполне стандартной. Но восстановительное элиминирование даёт теперь каталитически активный комплекс Ru(0). Цикл замкнулся.
Представьте себе, что вам захотелось сделать кросс-сочетание с одной стороны с C-H субстратом, и с другой стороны, например, с бороновой кислотой. В чём проблема? Проблема в том, что оба этих соединения - нуклеофилы. И мы знаем, что в таких случаях просто реакцию этих двух соединений уравнять невозможно - нужно куда-то деть два электрона, или, что то же самое, гидрид. Да и с остатком борной кислоты проблема - его тоже нужно пристроить.
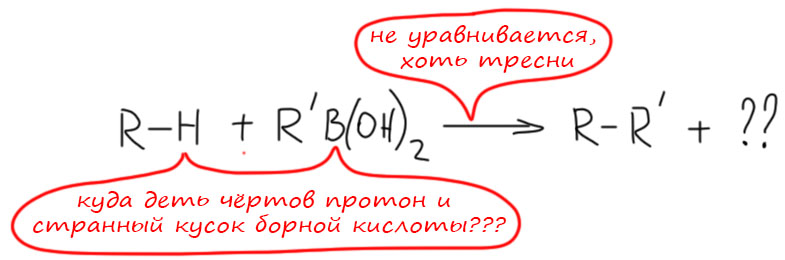
Всё тот же Какиути с сотрудниками придумал весьма неочевидный способ сразу пристроить и то, и то, и даже сделать реакцию каталитической, с добавлением ещё одного реагента, естественно (Kakiuchi, F. et al J. Org.Chem. 2007, 72, 3600). Вот какая простая система. Уже известный нам рутениевый пред-катализатор и два реагента. И вроде всё. Что там ещё - растворитель какой-то странный?
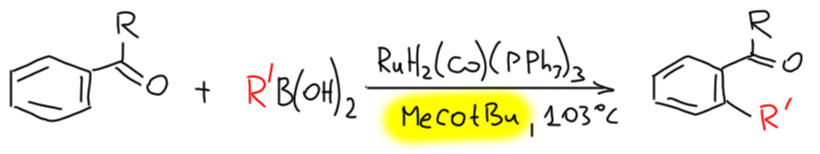
Пинаколин. Это ещё зачем? Наверное, просто вместо ацетона, но чтобы температура была побольше. Стоп, а куда же девались те куски, которые мы пытались пристроить?
И вот какая в этом цикле происходит занятная история. Начинается он так же, как и реакция кетона с алкеном - преактивация даёт комплекс Ru(0), который направленно окислительно присоединяется к связи C-H, образуя гидрид рутения. А вот дальше в дело вступает пинаколин - он внедряется по связи Ru-H (формально это просто гидридное восстановление карбонила, похоже на алюмогидрид и т.п.). Рутений всё же похож на палладий, ему тоже не очень нужен кислородный лиганд, а бору как раз он очень идёт - следует переметаллирование. Получается, что пинаколин в два захода оприходовал и бесхозный гидрид и бессмысленный остаток борной кислоты. Ну а дальше обычное восстановительное элиминирование, и дело в шляпе.
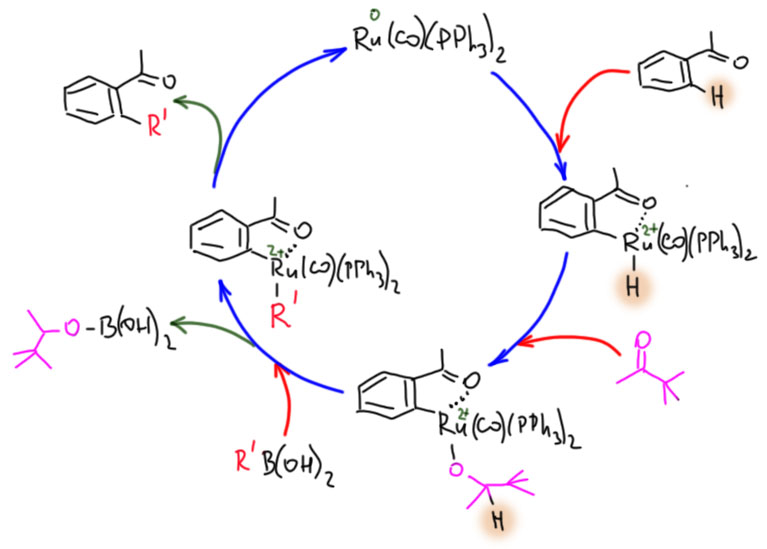
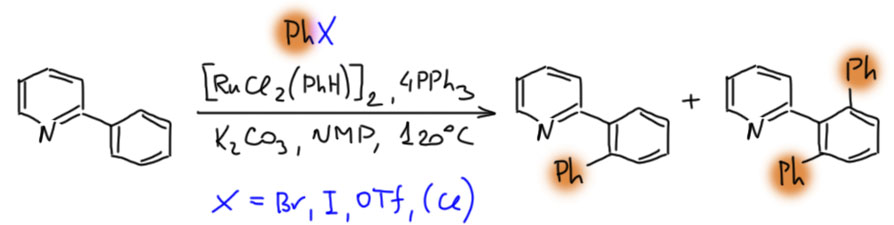
Совсем другой тип каталитической реакции с участием рутения открыли в самом начале нового века Сюити Ои и Ёсио Иноуэ. В присутствии бензольного комплекса Ru(2+) бензол с направляющей группой (2-фенилпиридин - это один из самых популярных субстратов для исследования реакций, включающих направленное металлирование) реагирует с стандартными ароматическими электрофилами - и это был первый пример прямого кросс-сочетания через C-H связь, с протоном, не имеющим повышенной кислотности.
В этом каталитическом цикле металлирование происходит Ru(2+), то есть так же, как мы рассматривали выше - в результате электрофильной активации. Координация по атому азота направляет металлирование. Так как это довольно ранняя работа, в ней еще нет хорошего представления о координационной сфере металла, и мы для простоты опустим остальные лиганды - всё равно точно непонятно, каковы они. Важна только степень окисления.
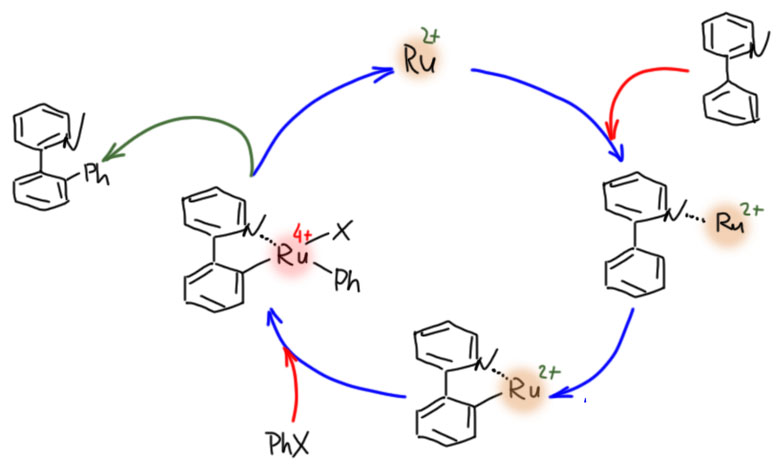
Пока что ничего нового относительно палладия нет. Палладий тоже отлично прометаллировал бы 2-фенилпиридин. Но дальше происходит то, о чём палладий может только мечтать, и о чём мечтал Бернард Шоу, когда грезил о реакции Хека и кросс-сочетании с участием Pd(4+). Для палладия это реализуется только с алкилгалогенидами, как в каскадах Каталлани, но никогда с ароматическими электрофилами.
А вот Ru(2+) делает это играючи и окислительно присоединяет ароматические галогенпроизводные и трифлаты. Дальше - обычное восстановительное элиминирование и возврат в цикл. Если есть ещё электрофил, в продукте остался ещё один атом водорода, который можно заместить.
Каталитический цикл Ои-Иноуэ, включающий направленное металлирование комплексом рутения (2+) с последующим превращением рутенацикла (некоторые предпочитают более короткое рутацикл, что нам, конечно, должно быть глубоко противно, потому что название металла рутений происходит от средневекового латинского названия нашей страны Ruthenia и уполовинивать его - не позволим!) в продукты, оказался чрезвычайно плодотворным. Эти реакции всегда используют направленное рутенирование, и поэтому во множестве исследуют самые разные направляющие группы - разные гетероциклы, функциональные группы, заместители, в том числе такие, которые вводят специально для этой цели а затем удаляют.
В реакциях этого типа пробовали много разных пред-катализаторов, но безусловным лидером стал ареновый комплекс рутения, изоструктурный с бензольным, использованным в первой работе Ои и Иноуэ, но вместо бензола используется довольно неожиданное соединение - кумол (p-cymene). Почему? Возможно, не в последнюю очередь потому что очень приятно пахнет хвоей. Но и потому, что оказался очень удачным анциллярным лигандом - он очень прочно держится на рутении по причине высокой донорности, и создаёт то самое стерическое напряжение в координационной сфере, заставляющее реакции каталитического цикла шустро бежать вперёд, накручивая круги в TON.

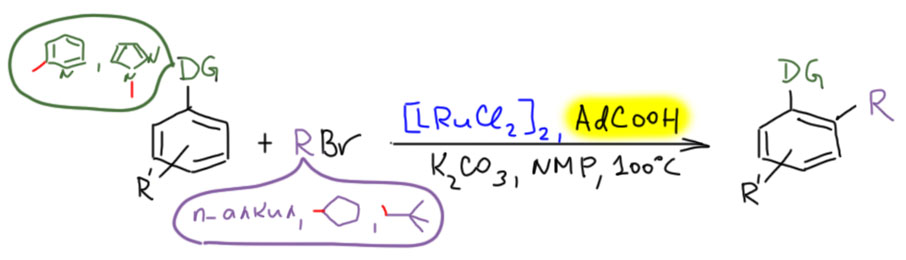
Например, один из самых ревностных чемпионов химии рутения, Лутц Акерманн сделал алкилирование без перегруппировок алкила (Ackermann, L. et al Angew. Chem., Int. Ed. 2009, 48, 6045).
Обратите внимание на странную добавку - 1-адамантанкарбоновую кислоту. Это тоже типичная вещь в этой химии - добавка такой кислоты, точнее, ее соли (там же избыток основания ещё), создаёт карбоксилатный лиганд на рутении, участвующий в стадии направленного C-H рутенирования. А стерический объём действует в том же направлении, что и объёмистый анциллярный лиганд кумол.
Тецуя Сато и Масахиро Миура с сотр. впервые сделали и рутениевый вариант реакции Фудзивары-Хека, использовав направляющие свойства карбоксильной группы, и высокодонорные пятичленные гетероциклы.
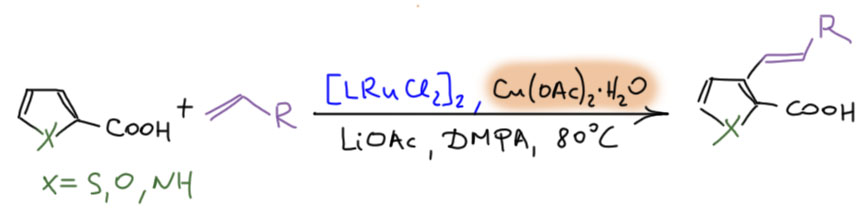
Ueyama, T.; Mochida, S.; Fukutani, T.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2011, 13, 706.
Металлы и их комплексы участвуют и во вполне обычных реакциях, хорошо известных в обычной органической химии, принося в них свои специфические свойства и умения. Одна из таких реакций, весьма популярных в современном синтезе – алкилирование по Фриделю-Крафтсу, типичная электрофильная активация C-H связей. Глянем-ка, что сюда внесли переходные металлы. Что нового увидим на страницах глянцевых журналов с заоблачным импактом.
Реакция Фриделя-Крафтса, точнее алкилирование по Фриделю-Крафтсу, в классической органической химии немыслима без безводного хлористого алюминия. Мы все знаем ее капризный нрав.
Но в новом 21-ом столетии нельзя жить воспоминаниями о 19-ом. Нужно идти вперёд. Мы знаем куда - экономия атомов, катализ, селективность, асимметрический синтез, бла-бла-бла, всё такое...
Посмотрим, что нам даёт новое время вместо безводного хлористого алюминия.
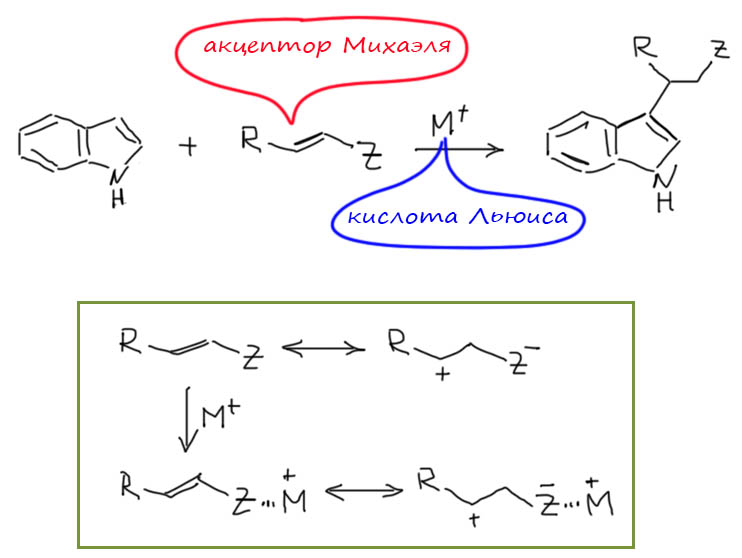
Как водится в новом времени, начинаем с лукавства. Нет, мы не будем метилировать бензол и тому подобное. Мы возьмём очень-очень донорное ароматическое соединение, пиррол или индол, и попробуем его алкилировать чем-нибудь очень мягким, слегка электрофильным, но таким, что можно дополнительно активировать. Это понятно, электрофильность активируется кислотностью Льюиса, и у комплексов металлов этого свойства не занимать. Чтобы можно было активировать, возьмём обычные акцепторы Михаэля типа акрилатов или акрилонитрила - в таких молекулах за кислород или азот можно зацепить кислотой Льюиса, что и повысит электрофильность. А почему мы называем это алкилированием по Фриделю-Крафтсу? Алкилированием - потому что к ароматическому кольцу присоединяется насыщенный атом углерода, а уж что там ещё, нам не важно. Фриделя-Крафтса потому что электрофил активируется кислотой Льюиса. Но ведь это типичная реакция Михаэля, мы на 3 курсе проходили? Да, одновременно это и Михаэль, но иногда в такой реакции Фриделя-Крафтса используют и карбонильные соединения, и имины и другие электрофилы. Поэтому Фридель-Крафтс - более общее название.
А что мы хотим добиться? Конечно, стереоселективности, вещи немыслимой в обычном Фриделе-Крафтсе. Но в замещённых акцепторах Михаэля есть прохиральный атом, и именно тот, который и является электрофильным центром. Игра стоит свеч.
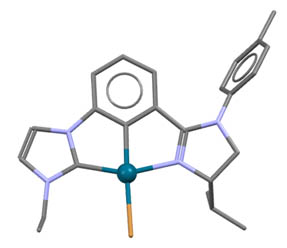
Вопреки обыкновению не будем копаться в истории этого метода, а посмотрим сразу на один из последних писков моды, чтобы и энантиоселективность заоблачная, и пинцер, и вообще всё с иголочки. Достижение Сон Маопина с сотрудниками из китайского города Чжэнчжоу очень показательно для современного состояния этой области. Кислотой Льюиса здесь служит весьма нетривиальный CCN-пинцер, в котором есть и NHC-карбен, и хиральный центр. Хиральный центр правда болтается где-то сбоку и эффективного переноса хиральности не обеспечивает, хотя на структуре видно, что асимметрия комплекса довольно близка к реакционному центру, но она невелика и состоит всего-навсего в том, что с одной стороны плоской молекулы куцый изопропил, а с другой водород. Надо было раскошелиться на что-нибудь пообъёмистей. И здесь используется немного другой электрофил, не акцептор Михаэля, а имин, но он тоже неплохо активируется кислотой Льюиса.
Song M-P. et al, Organometallics, 2018, 37, 2325–2334
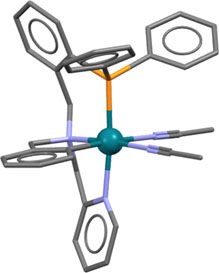
А вот еще более свежая работа по энантиоселективной реакции Фриделя-Крафтса-Михаэля, и в этой работе используется настоящая диковина - хиральный катализатор, хиральность которого обусловлена не лигандами, а стереогенным атомом родия (хотя внимательный глаз усмотрит еще и хиральность в самом лиганде, имеющем стереогенный азот в закреплённой хелатированием конфигурации). Этот комплекс вполне эффективно катализирует алкилирование индола нитроолефином.
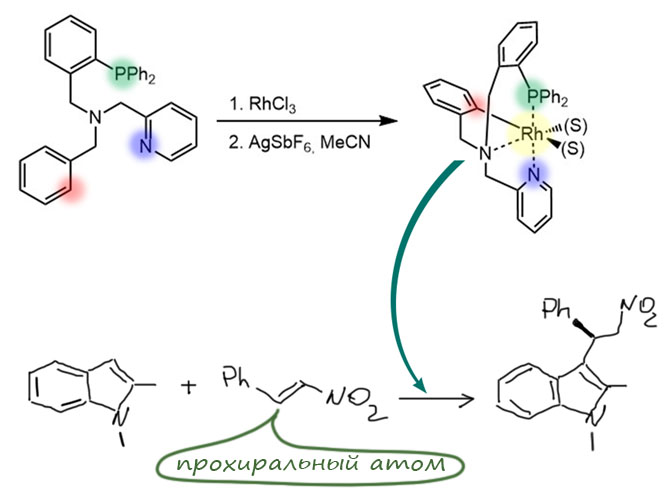
Carmona M et al, Organometallics, 2019, 38, 988
(S) - это растворитель, ацетонитрил
Достижения CH-активации не ограничиваются методами соединения различных углеродных фрагментов, но были открыты и разнообразные и иногда довольно удобные методы введения других групп и заместителей. Особенно важен метод CH-борилирования, ставший невероятно популярным в синтезе из-за удобства и надежности для прямого введения в ароматические кольца борных заместителей далее используемых в кросс-сочетании по Судзуки-Мияуре, и позволивший дотянуться с кросс-сочетанием до самых скрытых мест в больших органических молекулах, для которых невозможно или очень затруднено применение классического кросс-сочетания через введение подходящих уходящих групп.
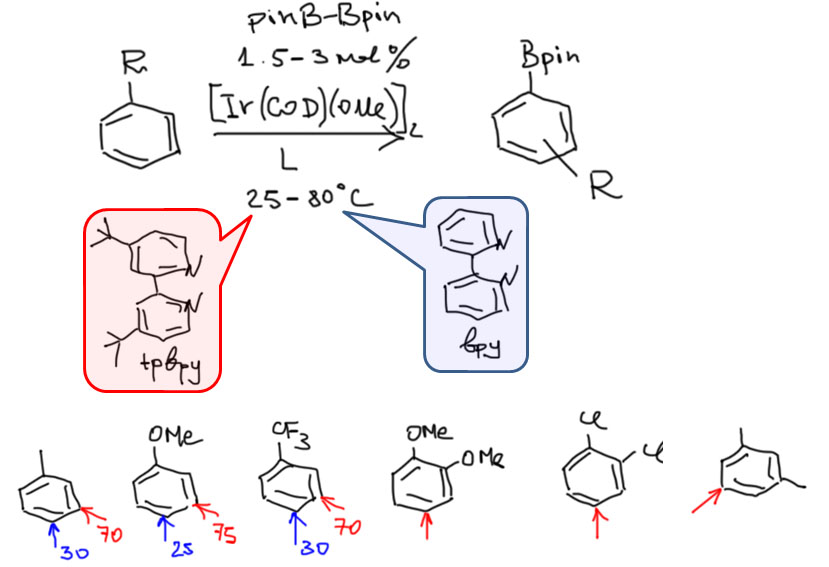
Метод получения арилборных кислот (в виде пинаконовых эфиров) без необходимости создавать галоген или другую уходящую группу стало важнейшим пополнением и без того огромного набора методов, позволяющих получать борорганические соединения для кросс-сочетания по Судзуке-Мияуре. Метод был разработан в результате впечатляющего штурма проблемы сразу тремя группами Хартвига, Исиямы и Мияуры, и Смита Третьего-го в 2001-2002 г. Все они, работая независимо и ревностно следя за успехами конкурентов, пришли к очень близким выводам,
Реакция идет в присутствии специфического иридиевого катализатора в очень мягких условиях, и весьма легко дает самые разнообразные боронаты из производных бензола и гетероциклических соединений. Реакция отличается необычной региоселективностью и дает преимущественно мета-изомеры из монозамещенных бензола, как донорных, так и акцепторных. В ди- и более замещенных бензолах место борилирования однозначно определяется минимизацией стерических препятствий. Популярность этого метода в синтезе огромна - с помощью комбинации этого борилирования и кросс-сочетания получено огромное количество новых органических соединений.
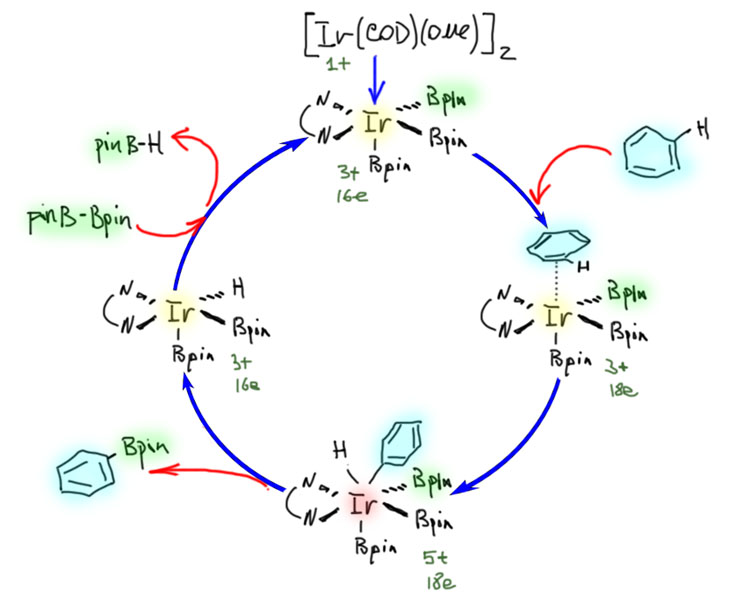
Цикл включает комплексы иридия в степенях окисления 3+ и 5+, хотя в реальности связь иридий-бор вряд ли поляризована в сторону бора, и это объясняет, почему иридий в формальной степени окисления 3+ охотно вступает в реакцию окислительного присоединения, и для чего в координационной сфере иридия такая пропасть борильных лигандов, пара из которых остается там все время и фактически просто создает избыточную электронную плотность на атоме металла. Необычная региоселективность реакции с монозамещенными производными бензола в рамках такого механизма объясняется минимизацией стерических препятсвий при образовании промежуточного η2-аренового комплекса, в котором и происходит окислительное присоединение C-H связи.