
Вспоминаем основы координационной химии
В этом курсе мы изучаем применение соединений переходных металлов в органической химии. Можно было бы из этого сделать вывод, что предметом курса является органическая химия, и мы просто добавим к известному арсеналу органических реакций новые реагенты на основе производных палладия, меди, иридия, молибдена, циркония и других переходных металлов, и будем, как это принято в органической химии, смотреть, как эти реагенты решают проблемы органического синтеза, какая у них селективность, диапазон применения, ограничения, и т.д. В классической органической химии именно так: главные задачи – синтез новых органических молекул, усовершенствование синтеза известных молекул. Поэтому и подход такой, ориентированный на синтез. Даже когда мы в органической химии разбираем механизмы, то в основном только для того, чтобы лучше понять синтетические возможности реакций.
Но в разделе органической химии, основанном на применении производных переходных металлов, этот подход не работает. Проблема в невероятном разнообразии уже введенных в синтез реагентов и катализаторов этого типа. Все известные d-элементы, представленные в Периодической таблице, кроме короткоживущего радиоактивного технеция, задействованы в органическом синтезе десятками и сотнями комплексов разной степени сложности, причем речь идет не об экзотических разработках, опубликованных в одной статье и положенных на полку, а на реально применяемых методах синтеза с десятками-сотнями-тысячами примеров применения. И мы принципиально оставляем за пределами данного курса f-элементы, очень интересные и перспективные, но весьма специфические и требующие отдельного рассмотрения.
Производные переходных металлов – реагенты и катализаторы – необычны для органиков. Это комплексные соединения, в состав которых входят разнообразные лиганды, от самых простых типа хлорида или пиридина, до ужасающе сложных и, на первый взгляд, совершенно непонятных, ощетинившихся объемистыми замеcтителями многоэтажных фосфинов, гетероциклических карбенов, макроциклов и т.д. Все эти лиганды – не бессмысленные украшения, типа, кто придумает сложнее и рогатее, тот и герой, – а строго определенные по функциям неотъемлемые части реагентов и катализаторов, определяющие селективность, каталитическую активность, реакционную способность, препаративный диапазон, стабильность и тому подобные важнейшие характеристики. Замена одного лиганда на другой часто приводит к драматическому изменению свойств, а сами лиганды подбирают под почти каждую специфическую решаемую задачу. Нет никаких универсальных рецептов, общих методов, рекомендованных методик. Любой химик, планирующий применять комплексы переходных металлов для решения своей задачи, должен понимать, как все это работает, или рискует без пользы потерять кучу времени и денег и намертво разочароваться.
Единственный надежный ключ к этому богатству – координационная химия, химия комплексных соединений. Несмотря на то, что большинство функциональных лигандов – чисто органические соединения, а воспринимаемая невооруженным взглядом сложность, навороченность катализатора или реагента часто сидит в органической части комплекса, эта наука традиционно примыкает к неорганической химии, а не к органической. Степень оскорбления правоверного органика еще усугубляется тем, что так называемые металлоорганические соединения, с точки зрения современной химии, это просто одна из разновидностей комплексных соединений металлов, а следовательно законный предмет изучения координационной и неорганической химии. В этом несложно убедиться, заглянув в свежий или не очень свежий номер любого из известных неорганических журналов с хорошей и долгой историей: Inorganic Chemistry, Dalton Transactions, Inorganica Chimica Acta, European Journal of Inorganic Chemistry – в глазах будет рябить от явно органических молекул. Но методология, использованная в статьях, будет другой, непривычной органикам – это терминология и методология координационной химии, которая крутится вокруг металла, а лиганды рассматривает не с точки зрения их органической природы, а скорее как управляющие элементы, настраивающие центральный металл на исполнение какой-то важной роли.
Поэтому и мы тоже углубимся в предмет с этой стороны. Нас вообще не будет интересовать, откуда берутся те или иные лиганды и как их синтезируют, но будет интересовать, как устроена химия комплексов, что в ней определяется центральным металлом, а что лигандами. И начнем мы с того, что просто вспомним то, что наверняка проходили хотя бы на 1 курсе, а возможно и в рамках каких-то спецкурсов, – с основ координационной химии. Многие все это и так знают наизусть и с самого раннего детства – ну, тогда просто пропустите, или даже полистайте, возможно, что-то все-таки забыли, и будет полезно напомнить.
Сразу предупреждаю, что в данном курсе будет сведено к минимальному минимуму все, связанное с квантовой картиной химии, не будет даже теории кристаллического поля, не говоря уж о корректно нарисованных МО комплексов и результатов DFT расчетов. Все это очень важные и полезные вещи, и, начиная с некоторого уровня, без них не обойтись. Но практика показывает, что вполне удовлетворительное описание свойств и реакций комплексов переходных металлов с акцентом на использование в органической химии можно получить и без этой артиллерии. Может быть, потом добавим, если появится потребность. А пока ограничимся только самыми простыми сведениями о том, как устроены комплексы и связи.
И в этом и во всех других разделах информация будет устроена так: сначала некоторое подобие слайдов, приблизительно соответствующее лекционному материалу, и у многих слайдов будет кнопка, отсылающая к более подробному описанию. В конце появятся задачи на усвоение и понимание материала темы.
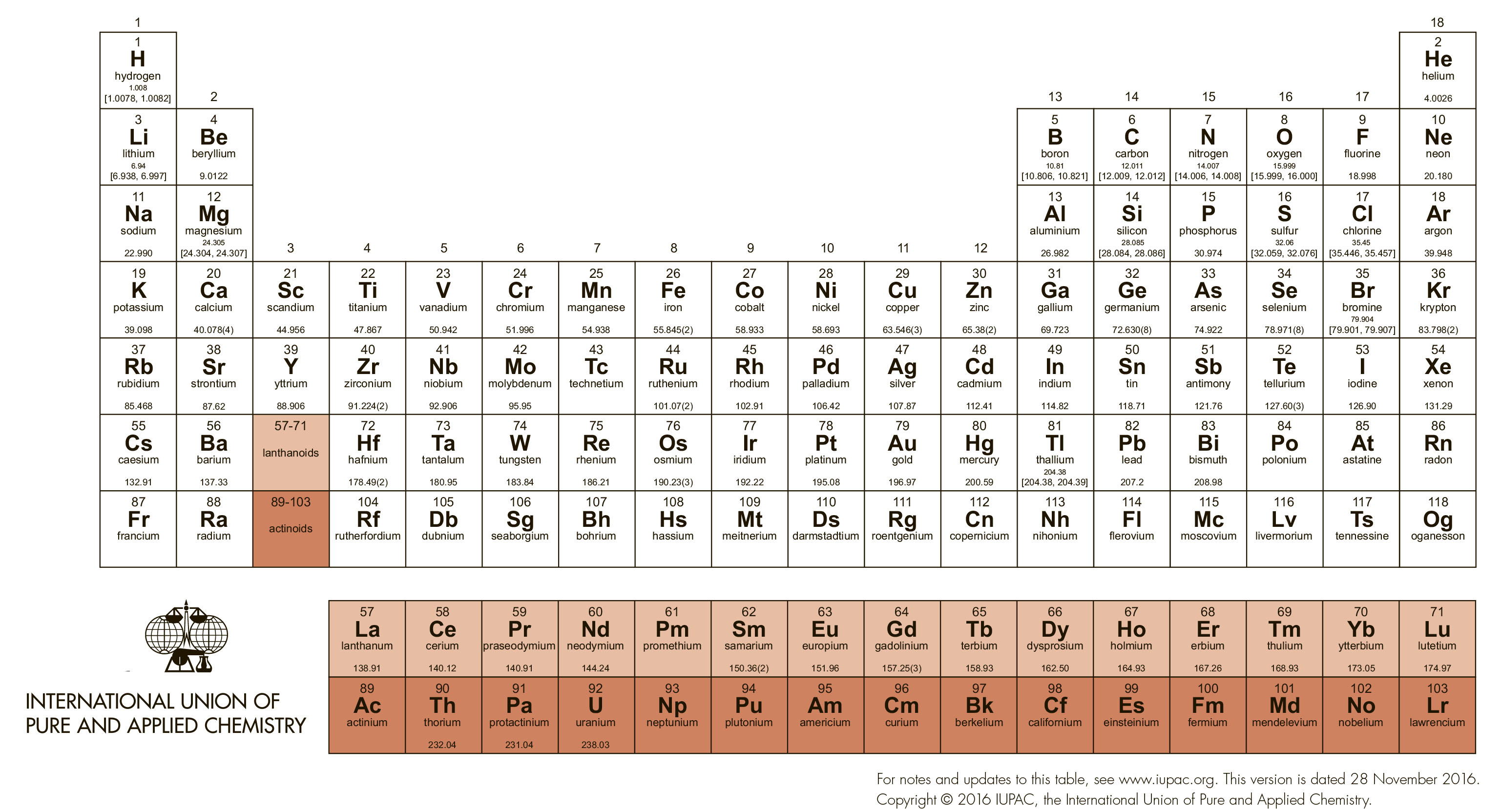
Будем пользоваться длиннопериодной формой Периодической таблицы элементов Менделеева, очень удобной потому, что положение элемента точно отражает последовательность заполнения валентных оболочек. Стандартизованная форма такой таблицы рекомендована ИЮПАК.
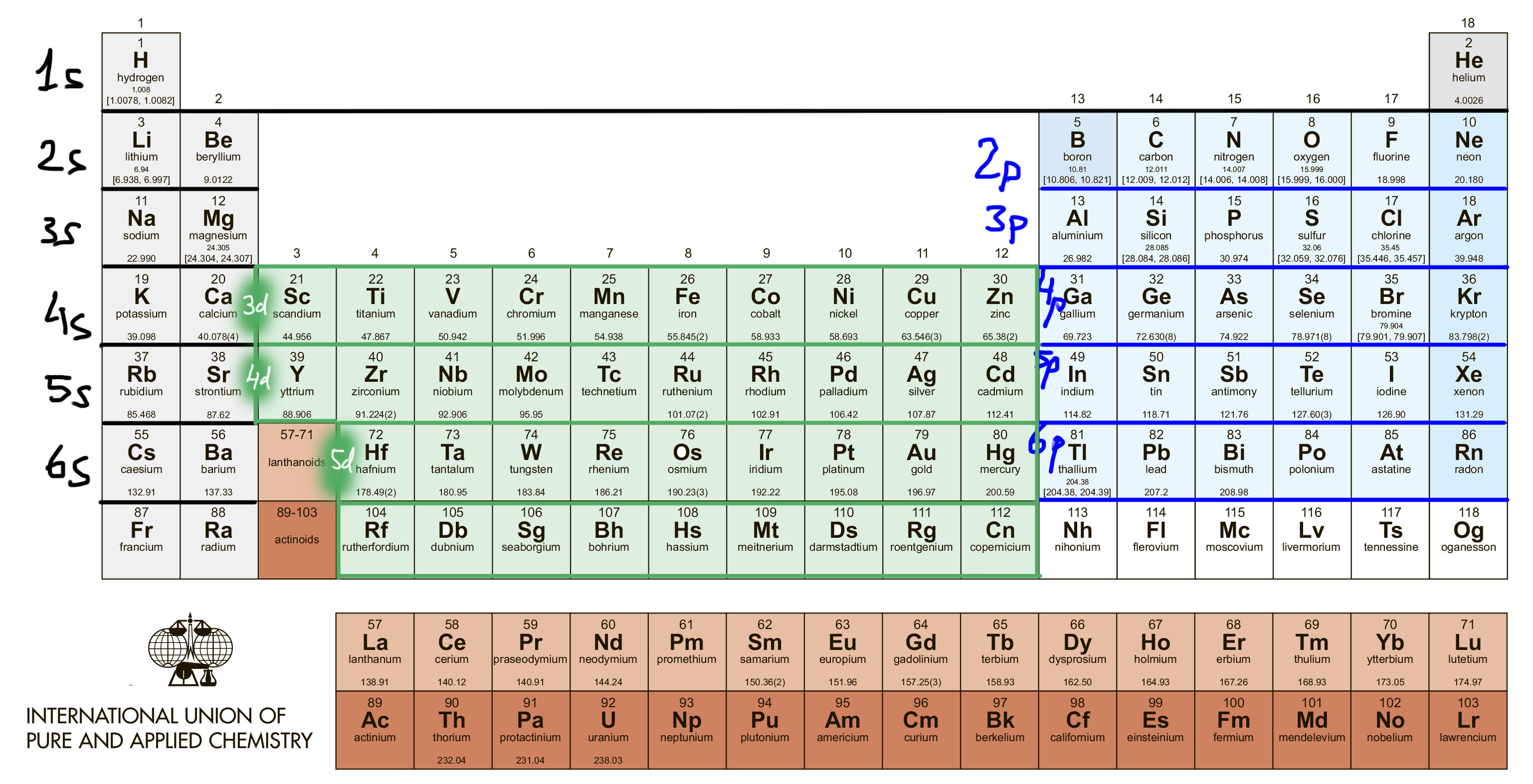
D-элементы в такой Таблице занимают место ровно в середине между s- и p-элементами в группах от 3 до 12, что соответствует постепенному заполнению d-оболочки, состоящей из 5 орбиталей и вмещающей 10 электронов. Такое положение отражает то, что очередной d-уровень по энергии находится между s и p уровнями следующей по номеру (значению главного квантового числа) оболочки
Три ряда d-элементов соответствуют заполнению 3d, 4d, и 5d-уровней. В координационной химии имеют дело с атомами металлов в окружении лигандов, взаимодействие лигандов с металлом изменяет порядок заполнения уровней. Количество d-электронов на атоме нульвалентного металла в комплексе соответствует номеру группы в длиннопериодной Таблице. Для двух последних групп в валентной оболочке появляются s-электроны
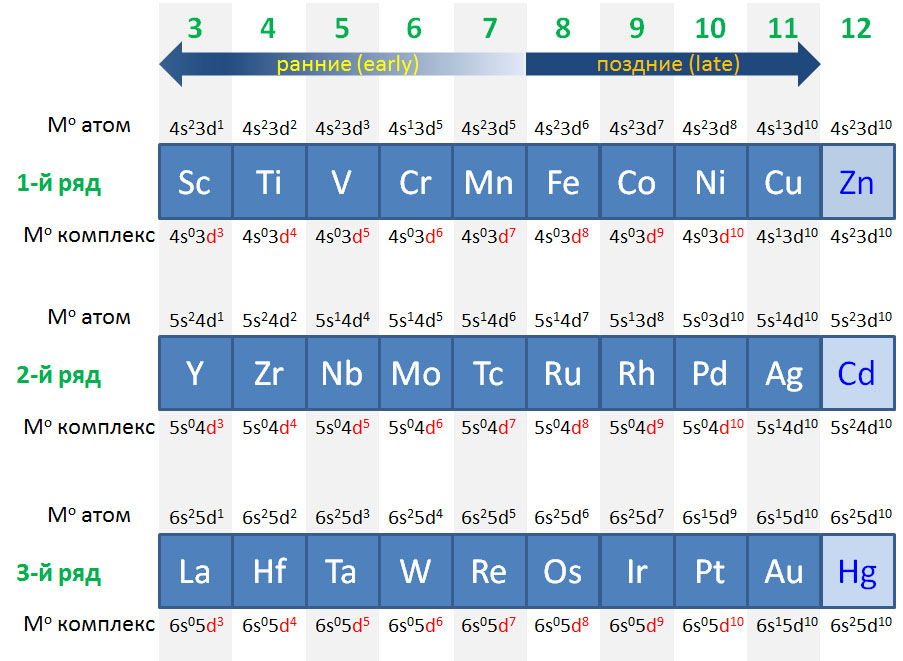
В комплексах металлы могут иметь разные степени окисления. Количество d-электронов на атомах металлов с положительной степенью окисления определяется разностью между номером группы и степенью окисления. Степень окисления не может быть больше номера группы, и не больше 8, поэтому конфигурация d0 возможна только для элементов первых восьми групп. У элементов 1-11 групп есть валентные состояния с частично заполненной d-оболочкой: эти элементы являются переходными металлами. Элементы 12 группы (цинк-кадмий-ртуть) переходными иметаллами не являются - d-оболочка всегда заполнена.
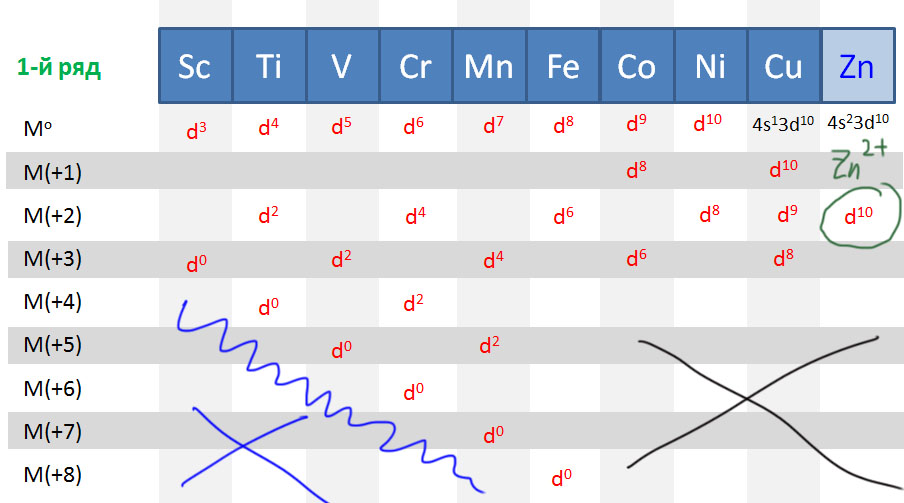
Переходные металлы делятся на ранние и поздние. Это деление достаточно условно, но отражает склонность ранних к образованию высоких степеней окисления и конфигурации d0, и вторых к образованию низковалентных состояний с конфигурациями d8 и d10, что имеет глубокие следствия в химии этих элементов. Элементы 11 группы (медь-серебро-золото) имеют много черт родства с поздними переходными металлами, но и очень специфические черты, сближающие их с s-металлами, поэтому их часто выделяют в особую группу переходных металлов.

Координационные, комплексные, металлоорганические соединения
Химия, которой мы будем заниматься, требует участия производных переходных металлов или как катализаторов, или как стехиометрических реагентов. Производные переходных металлов – не все вообще, но те, которые нас будут интересовать – всегда устроены по одной схеме: атом металла и вокруг него органические или неорганические лиганды. Такие соединения принято называть координационными соединениями. Есть ещё старый термин “комплексные соединения”, восходящий к работам Альфреда Вернера, получившего за это Нобелевскую премию в 1913. С нашей точки зрения разницы между этими терминами нет. Мы будем называть соединения переходных металлов и комплексами переходных металлов, и координационными соединениями переходных металлов. В более строгом смысле разница есть, и термин “координационные соединения” имеет более общий смысл.
Металлоорганическими соединениями называют координационные соединения, в структуре которых есть хотя бы одна связь металл-углерод, любого типа (ионного, координационного, ковалентного).
Простые лиганды
Теперь займемся лигандами. Простые лиганды связаны с атомом металла всегда через один атом, то есть в комплексе всегда просто и однозначно можно найти связи металл-углерод, металл-водород, металл-галоген, металл-кислород, металл-азот и т.п. какой бы громоздкой и сложной молекулой ни был бы сам лиганд.
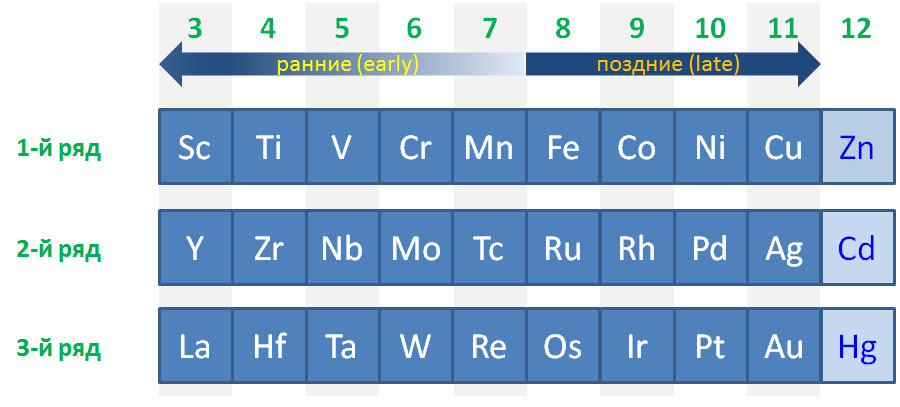
В координационных соединениях атом металла окружен лигандами - это так и называют: центральный атом металла и координационная сфера.
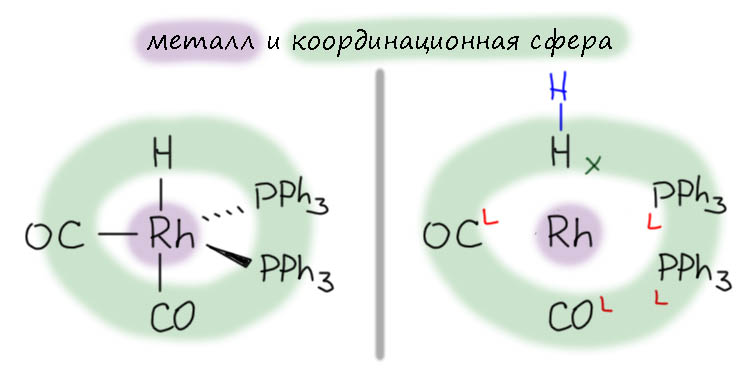
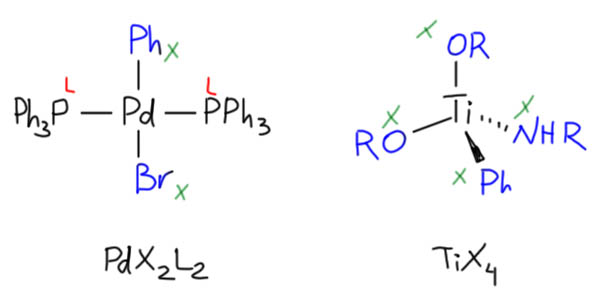
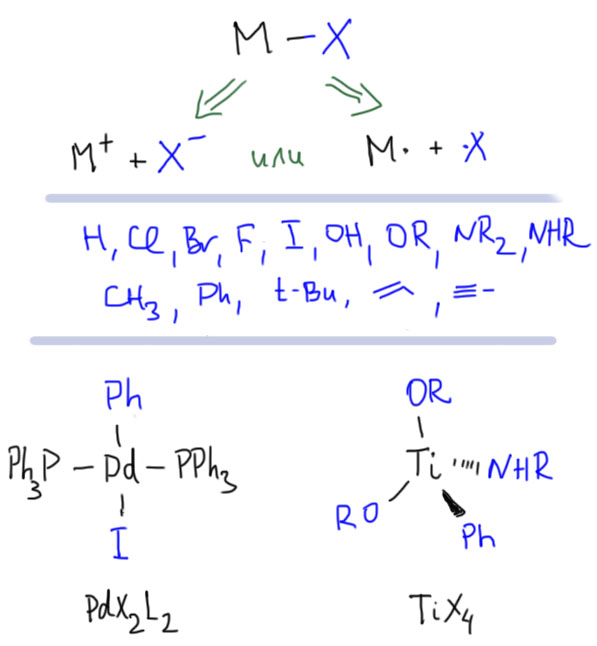
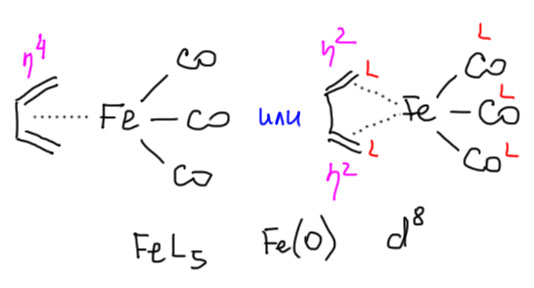
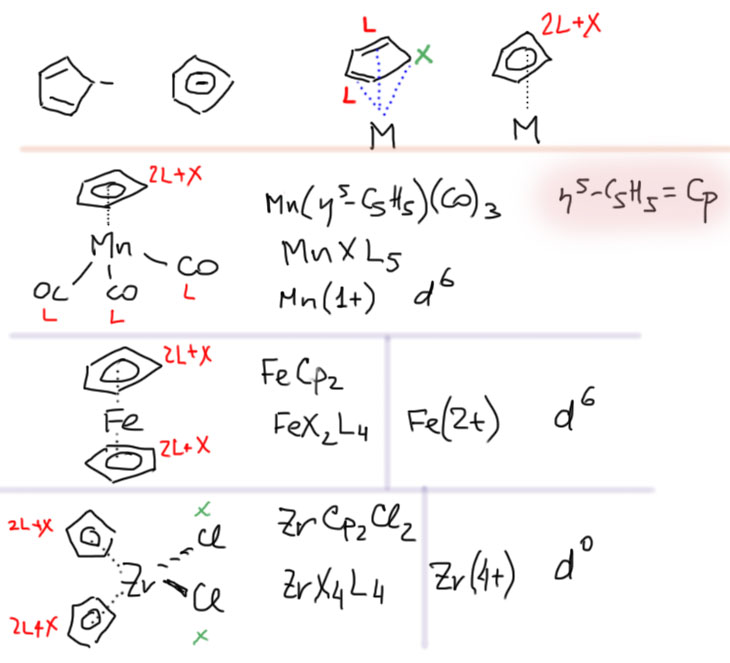
Сначала займемся простыми лигандами, связанными с атомом металла через один атом. Лиганды можно подразделить на X и L-типы. X-лиганды - одновалентные остатки молекул (можно мысленно добавить связь с атомом водорода), L-лиганды - нейтральные молекулы. Разобрав лиганды на типы, для любого комплекса можно написать обобщенную формулу, которая определяет структурный тип. В данном примере родий несет пять лигандов, один из которых (атом водорода, который можно представить, как часть молекулы водорода) - X-лиганд, остальные, молекулы фосфина и CO - L-лиганды, поэтому обобщенная формула будет MXL4.
Существует традиция обозначать связи с лигандами X-типа сплошной чертой, связи с лигандами L-типа пунктиром. Но это именно традиция, и в реальной научной литературе ей не очень аккуратно следуют. Поэтому лучше на эти обозначения не рассчитывать. Природа лиганда должна и может быть установлена однозначно без этой подсказки. Поэтому на этом сайте я не придерживаюсь этой негласной конвенции. Иногда это удобно, особенно в хелатных комплексах, но чаще это ничего не дает.
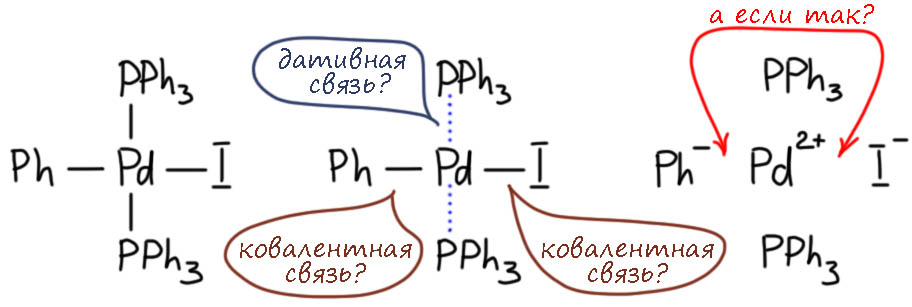
Действительно ли так просто отличить ковалентную связь от донорно-акцепторной? Или это просто две разновидности одного и того же?
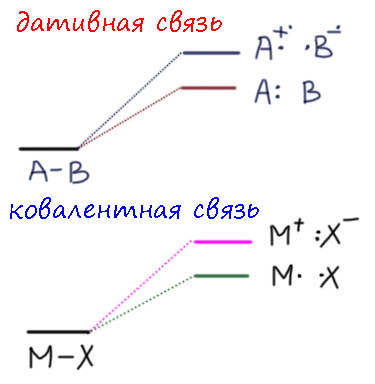
Ковалентная связь очень легко может быть представлена как донорно-акцепторная (дативная), если взять ион вместо атома металла, и анионные формы лигандов - собственно именно эту модель и использует одна из двух схем подсчёта валентных электронов металла. Были разные попытки формально определить разницу, например, критерий Халана (A. Haaland, Angew. Chem., Int. Ed. Engl., 1989, 28, 992–1007), ищущий разницу в путях минимальной энергии расщепления связей - гомолитическом или гетеролитическом. Но для переходных металлов это сделать малореально - у нас просто нет соответствующих надёжных термохимических данных.
Поэтому вместо классификации по типам связей комплексы лучше анализировать по типам лигандов, что установить намного проще и надёжнее.
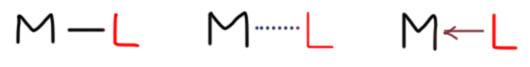
стрелка нежелательна
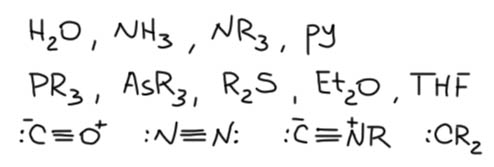
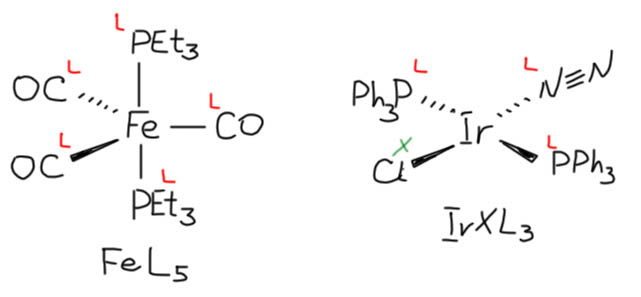
Лиганды L-типа образуют дативную (координационную) связь с металлом: металл предоставляет вакантную орбиталь (является кислотой Льюиса), а лиганд - неподеленную пару (является основанием Льюиса). В химии переходных металлов простыми L-лигандами могут служить почти любые молекулы, имеющие атом с неподеленной парой, даже такие, которые практически не проявляют ни основности, ни нуклеофильности в органической химии, например, молекулы CO и азота. И не обязательно устойчивые в свободном состоянии, например, карбены. Если в молекуле есть атом с неподеленной парой, будьте уверены, что найдется переходный металл, который захочет эту молекулу поиметь в виде L-лиганда, хотя при этом не обязательно образуются стабильные комплексы, а ставшая лигандом молекула изменит свою реакционную способность. На этом основано применение комплексов металлов для электрофильной (кислотной) активации молекул. Стоит отметить, что этот тип активации присущ не только переходным, но и непереходным металлам, хотя диапазон превращений, вызываемый такой активацией у переходных металлов гораздо шире.
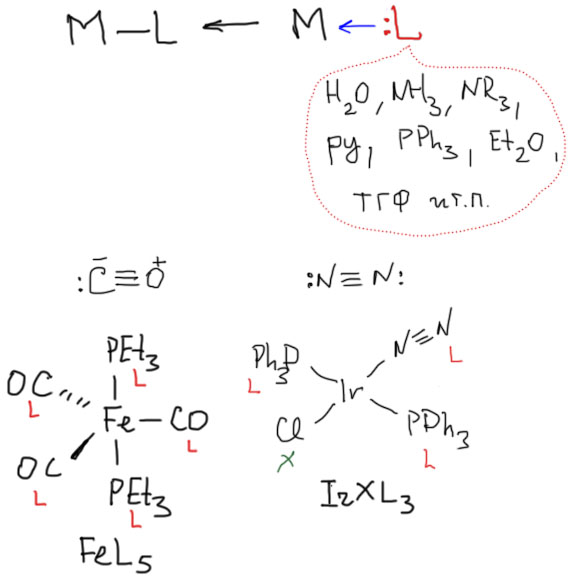
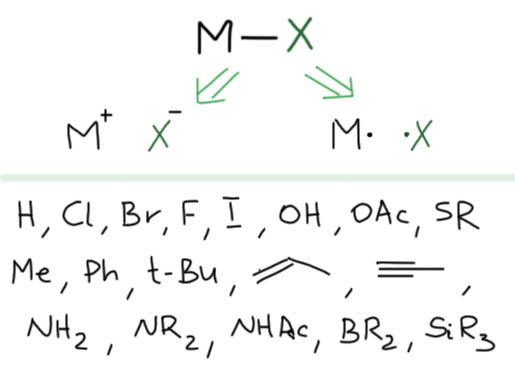
Лиганды X-типа образуют ковалентную связь с металлом, то есть можно представить гомолитический разрыв такой связи, образующий свободнорадикальную форму лиганда и металл с нечетным числом электронов в оболочке. Альтернативный способ образования такой связи - пара лигандов от анионной формы лиганда и вакантная орбиталь на металле с формальным положительным зарядом, и в этом случае связь можно назвать типичной координационной. Эта двусмысленность чисто формальна, и на нее можно не обращать внимания. Главный признак X-лиганда то, что если его мысленно отделить от металла, получится не устойчивая молекула, а одновалентный радикал или анион.
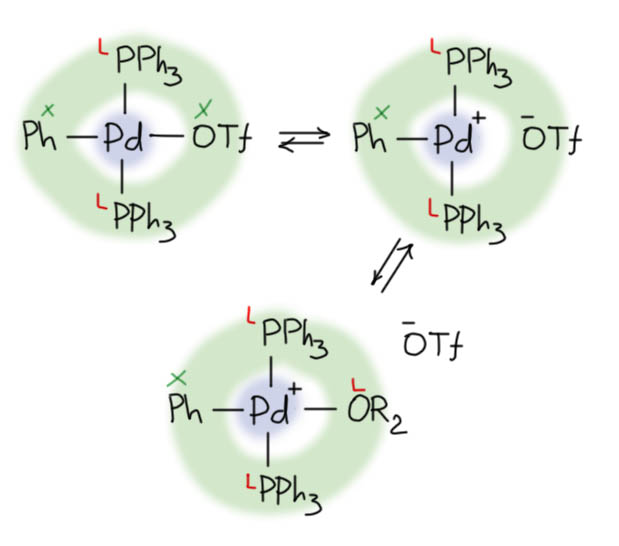
Анион любой кислоты может быть лигандом X-типа. Но есть анионы очень сильных кислот, которые обычно называют слабо-координирующимися анионами (WCA). Один из таких очень популярен в любой химии - трифлат. И вот вопрос - а он держится в координационной сфере металлов? Или образует ионные связи?
Бывает и так, и так. Часто держится и служит довольно обычным, хотя и лабильным X-лигандом. А иногда нет - и тогда уходит из координационной сферы (часто говорят: из внутренней сферы во внешнюю), а его место может занять другой лиганд, например, растворитель. Или даже что-то нужное. Этот приём чрезвычайно важен в химии переходных металлов.
Кроме трифлата есть и другие такие лиганды-нелиганды, но это уже экзотика.
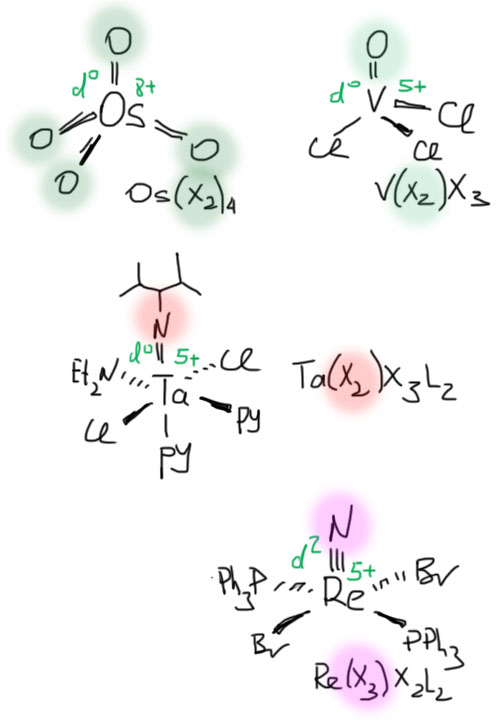
Есть ряд лигандов, которые, занимая одно место в координационной сфере металла, вносят вклад +2 или +3 в степень окисления металла, то есть ведут себя в этом отношении как два или три X-лиганда. Такие лиганды можно выделить в особый тип или подтип X2 и X3-лигандов. Типичным и даже архетипичным представителем X2-лигандов является оксо-лиганд, хорошо известный по таким простым координационным соединениям, как тетраоксид осмия, перманганат и хромат-ионы, соединения ванадила и других оксокатионов.
Другие лиганды X2-типа - имидо-комплексы, имеющие возможности более широкой вариации структуры за счет заместителя на азоте.
X3-лиганды представлены в основном нитридным лигандом в комплексах переходных металлов высокой степени окисления.
Углеродные лиганды X2 и X3-типов это карбены (но далеко не все) и карбины, с которыми мы встретимся при обсуждении метатезиса алкенов и алкинов.
Для определения степени окисления металла связи с лигандами X-типа (включая X2 и X3) всегда используют простое правило - неметалл получает минус, металл плюс. Это правило соблюдают всегда для всех неметаллов, включая водород. Степень окисления - формальная характеристика, которая не обязана отражать реальное направление поляризации связей. Гидридные комплексы металлов могут быть как гидридами, так и протонными кислотами, или даже совмещать эти два типа реакционной способности. Многие переходные металлы, особенно поздние, имеют немаленькую электроотрицательность.

Определяем степень окисления металла и электронную конфигурацию. Распределив лиганды по L и X-типам, и посчитав количество лигандов каждого типа, определяем степень окисления как число X-лигандов, L-лиганды в степень окисления вклада не вносят. Если у комплекса есть заряд, то прибавляем его к числу X-лигандов (с учетом знака).
Число d-электронов равно номеру группы минус степень окисления.

Простые (и не только) лиганды можно соединять в одной молекуле, собирая их с помощью более или менее гибкого органического (не обязательно!) фрагмента. Такие молекулы с несколькими координационными центрами называют полидентатными лигандами (бидентатными, тридентатными и т.д.). Такой большой многоцентровой лиганд может связываться с одним атомом металла, образуя комплекс-хелат. Или с двумя и более атомами металла, образуя многоядерный комплекс или кластер. Комплексы хелатного типа, а следовательно и сами такие лиганды играют колоссальную роль в органических реакциях, а придумывание таких лигандов и комплексов составляет одну из основных форм деятельности в данной области. Для поднятия самооценки этот в значительной степени хаотический мыслительно-экспериментальный или экспериментально-мыслительный процесс любят называть дизайном, намекая на строго целенаправленный характер этой деятельности.
Простые лиганды как X, так и L-типов могут соединяться по два, три, и более в одной большой молекуле, образуя бидентатный, тридентатный, … полидентатный лиганд. Если при связи такого лиганда с металлом образуется цикл, такой комплекс (а также сам цикл) называются хелатными, а лиганды называются хелатирующими или хелаторами. В одном комплексе может быть один или несколько хелатных циклов. Подсчет степени окисления, числа d-электронов делается точно так же, как в комплексах с более простыми лигандами, только отдельно считают каждый лигандный центр, помечая его как отдельный лиганд.
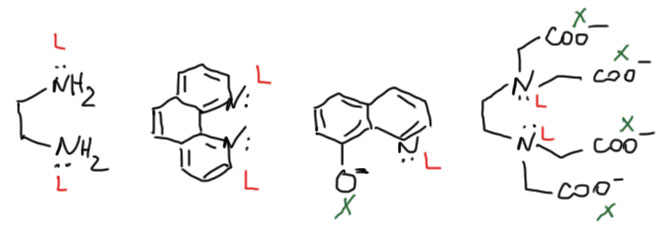
В реакциях с участием комплексов переходных металлов хелатирующие лиганды играют особую и совершенно колоссальную роль по множеству причин.
Хелатные лиганды прочнее держатся в координационной сфере, позволяя контролировать структуру комплекса, стереохимию, защищать координационную сферу от нежелательных лигандом и т.п. Хелатные комплексы гораздо устойчивее в условиях реальных реакций, и это очень важно особенно в каталитических реакциях, когда комплекс должен много раз проходить многостадийный процесс, не теряя важных компонентов координационной сферы металла. Если это не соблюдается, катализатор теряет активность (деактивируется), реакционную способность и селективность.
Хелатные лиганды, в другой стороны, имеют важный недостаток в тех же каталитических реакциях - они иногда занимают слишком много места в координационной сфере, а это ограничивает их возможности в одних реакциях, и совсем недопустимо - в некоторых других.
Из-за этого хелаторы не являются универсальным ответом на все проблемы катализа, и монодентатные лиганды не теряют своего значения. Химия - всегда искусство компромисса среди множества противодействующих факторов.
Как и в обычной органической химии самыми распространёнными циклами и в хелатных комплексах являются пяти- и шестичленные. Так же как и там основной причиной этого является компромисс между энергией образования (циклизации) - это всегда выгодно - и энтропийным фактором - это наименее невыгодно. Циклы такого размера не имеют существенных напряжений. Лигандов, образующих хелаты с такими размерами циклов невероятно много среди всех классов органических и элементоорганических соединений. А комплексы с такими хелатными циклами охотно образуют все металлы без исключения, во всех валентных состояниях.
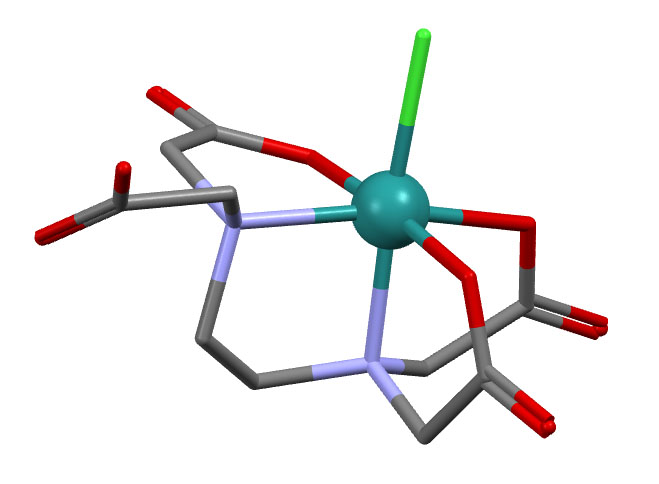
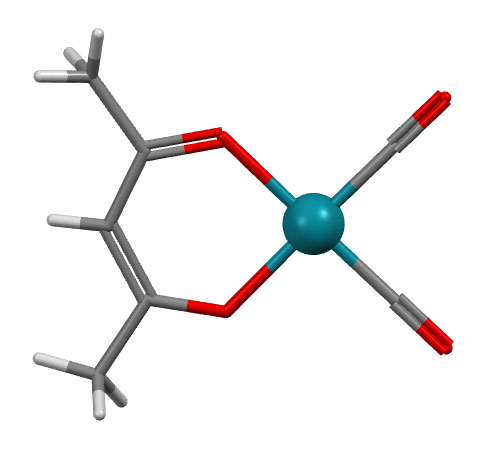
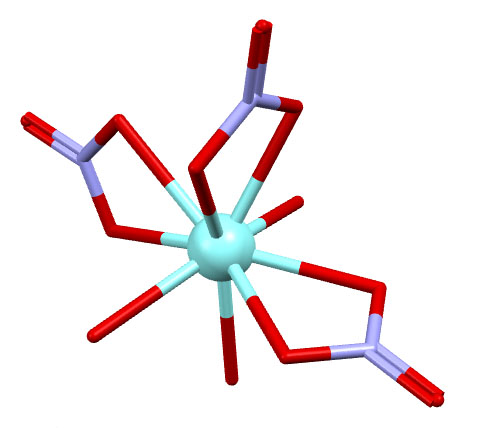
Но, в отличие от обычной органической химии, в химии переходных металлов другие циклы образуются тоже достаточно неплохо. Вот, четырёхчленный цикл в органической химии - один из самых трудных. Четырёхчленные циклы из атомов углерода страдают сильным угловым напряжением и энергетически невыгодны. В химии переходных металлов 4-членный хелатный цикл проблемой не является, и такие циклы встречаются достаточно часто. Многие простые анионы кислот - ацетат и другие карбоксилаты, карбонат, нитрат, сульфонат - очень часто в комплексах имеют именно такой способ связывания в комплексах. Очевидными причинами этого являются совсем другие стереохимические способности атомов переходных металлов. В октаэдрической конфигурации и всех производных от неё валентный угол в 90 градусов является нормальным и никакого напряжения не вызывает.
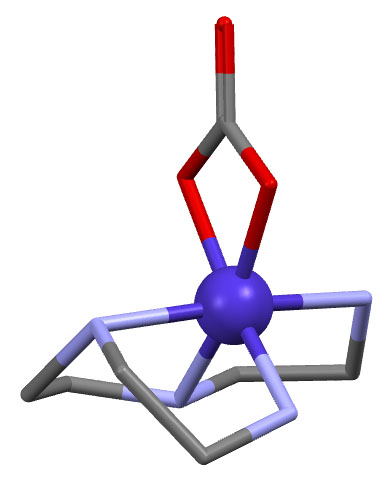
Бидентатные (и полидентатные, естественно) лиганды могут быть и хелаторами, сидя на одном атоме металла, и мостиковыми лигандами, образуя комплексы с двумя и более атомами металла. Мостиковое связывание обозначается греческой буквой μ (мю), поэтому и лиганды такие называют иногда мю-лигандами, впрочем, это просто синоним слова мостиковый (bridge, bridging). Комплексы с несколькими атомами металла, связанными мостиковыми лигандами называются многоядерными (multinuclear). Есть ещё понятие "кластер (cluster)", которое часто используют наряду с понятием "многоядерный комплекс", но в более аккуратном смысле "кластер" - более специфический тип многоядерного комплекса, содержащий прямые связи металл-металл.
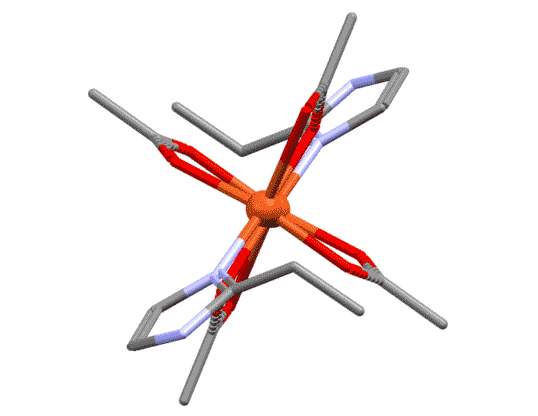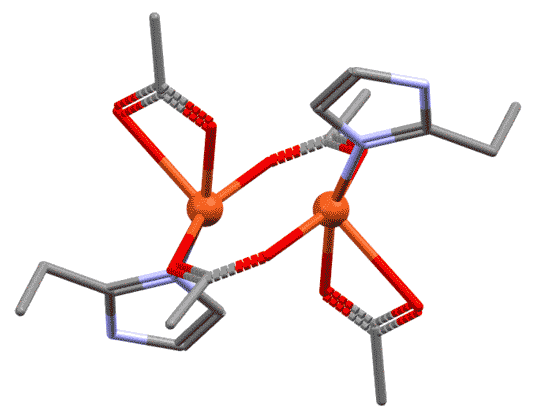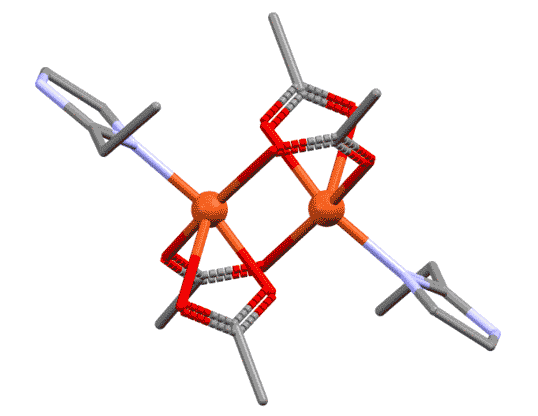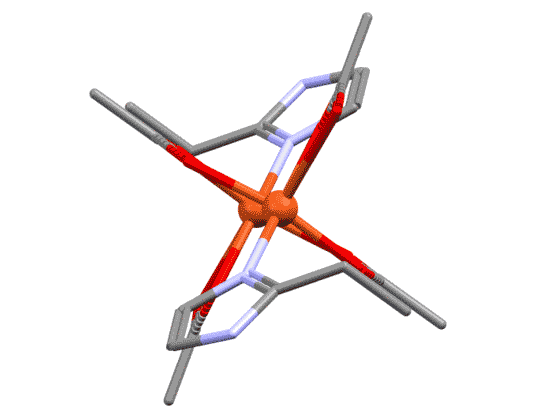
Мостиковыми чаще бывают бидентатные лиганды, образующие неоптимальные по размеру хелатные циклы (не 5- и 6-членные), например 4-членные, или, наоборот большие. Ацетат-ион, например, очень часто работает, как мостиковый лиганд, и такие комплексы очень широко растпространены. А вот, слева, пример биядерного комплекса меди (но не кластера!), в котором есть ацетатные лиганды сразу двух типов: и μ- и хелатных.

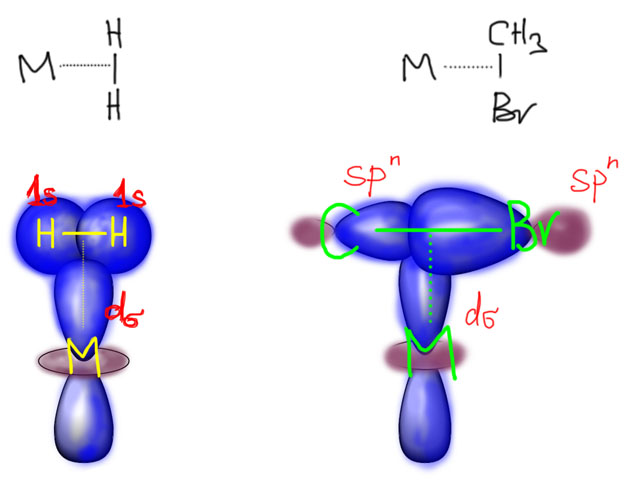
Даже самая простая ковалентная связь между атомами водорода в молекуле может быть использована переходным металлом для связывания. Другие σ-связи тоже годятся. Такое связывание происходит за счёт взаимодействия занятой связывающей орбитали σ-связи с вакантной d-орбиталью металла, подходящей по симметрии. Результирующая связь также имеет σ-характер. Комплексы такого типа часто вполне устойчивы, но в других случаях образуются как интермедиаты в реакциях. Координация переходного металла по связи почти всегда приводит к её ослаблению (причину разберём позже), вследствие чего обычно малоактивная связь приобретает способность участвовать в реакциях.
Соль Цайзе K[PtCl3(C2H4)]·H2O

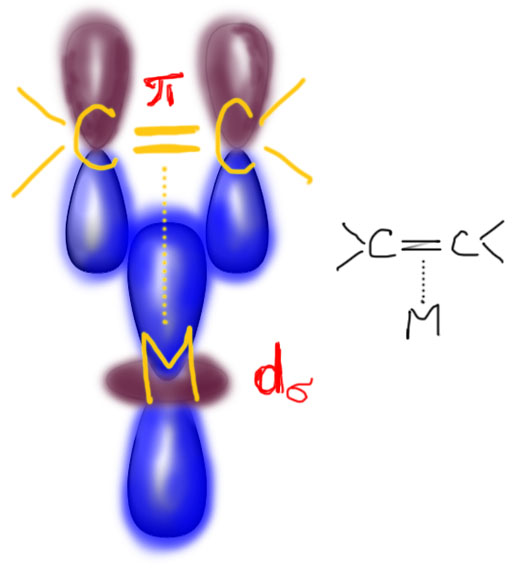
Очень легко образуют комплексы с переходными металлами олефины и ацетилены. Знаменитая соль Цайзе была получена почти 200 лет назад, считается первым металлоорганическим соединением. В этом случае донором является связывающая орбиталь π-связи (в ацетиленах каждая из π-связей может играть эту роль). Интересно, что природа связи олефина или ацетилена с металлом также имеет σ-характер, и для неё используется та же орбиталь металла dσ-типа, что и для образования комплексов с молекулами, не имеющими кратных связей. При этом, комплексы олефинов и ацетиленов традиционно называют π-комплексами, что иногда вызывает некоторую путаницу.
Структурно олефин связывается с металлом, как плоский фрагмент, перпендикулярный линии связи.
Лиганды, связываемые с металлом посредством своих π- или σ-связей, называются гапто-лигандами. В зависимости от того, сколько атомов сидит непосредственно на той связи, которая стала кординационным центром связывания, их маркируют как n-гапто, вместо слова "гапто" используют греческую букву η (эта). Водород, олефин, ацетилен - дигапто-лиганды (η2-лиганды), так как они цепляются за металл двухатомными связями. Это очень важно понимать - в таком комплексе не две связи металл-атом лиганда, а одна.
Нет ничего проще учёта лигандов такого типа в структуре. В подавляющем большинстве случаев это просто одиночные L-лиганды, занимающие одно место в координационной сфере (они вносят 1 в координационное число). Ацетилен, олефин, молекула водорода, что-то ещё - один L-лиганд.
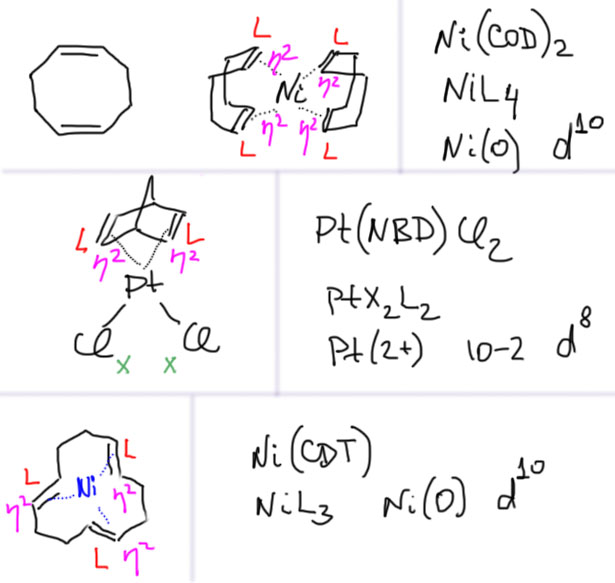
Гапто-лиганды, точно так же как простые лигандные центры могут быть в одной молекуле и образовывать хелатные комплексы с металлами. Среди таких гапто-хелаторов есть очень популярные в химии особенно поздних переходных металлов вспомогательные лиганды типа 1,5-циклооктадиена (COD), циклододекатриена (CDT), норборнадиена (NBD). Учёт таких хелаторов в координационной сфере металла ничем не отличается от учёта простых хелаторов.
Несопряжённые диены в комплексах работают как хелаторы. А сопряжённые? Чаще всего как единый лиганд, металл образует связи с занятыми орбиталями сопряжённой системы, которых, как известно, две. Связывание происходит в s-цис-конформации, если диен нежёсткий, а если жёсткий, циклический, то только с двойными связми, которые могут расположиться таким образом. Похоже на реакцию Дильса-Альдера, не правда ли?
Сопряжённый диен в комплексе является единым η4-лигандом. В анализе структуры комплекса можно было бы ввести новый тип четырёхэлектронного лиганда, но чтобы не плодить новые типы лигандов, поступают проще - учитывают как два L-лиганда. С ними всё получится правильно, но нельзя ни на мгновение забывать, что это не два независимых координационных центра.
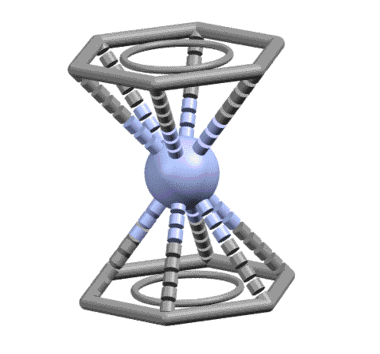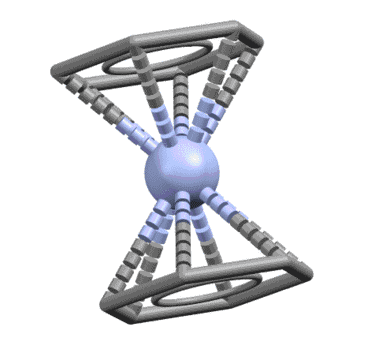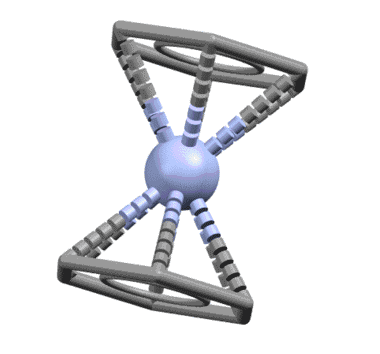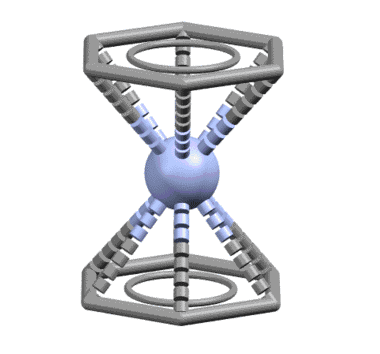
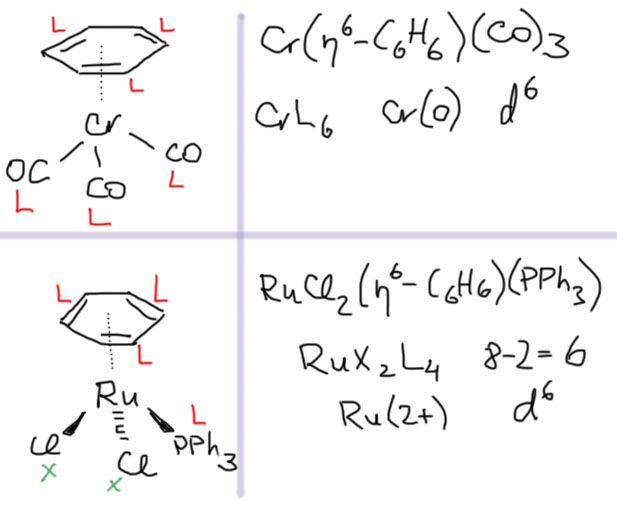
От сопряжённого диена один шаг до бензола, и шире, к ароматическому кольцу. Бензол ведёт себя как единый шестиэлектронный лиганд, связанный совершенно симметрично так, что расстояния от металла до каждого из шести атомов одинаковы. Комплекс бензола с нульвалентным хромом наряду с ферроценом послужил детонатором для взрывообразного роста металлоорганической химии во второй половине 20 века, что и было отмечено Нобелевской премией Уилкинсона и Фишера. При анализе комплексов бензол и другие ароматические соединения учитывают не как единый шестиэлектронный лиганд, а как три L-лиганда.
Известно довольно много комплексов, в которых бензолы связываются частично как η2- или η4-лиганды.
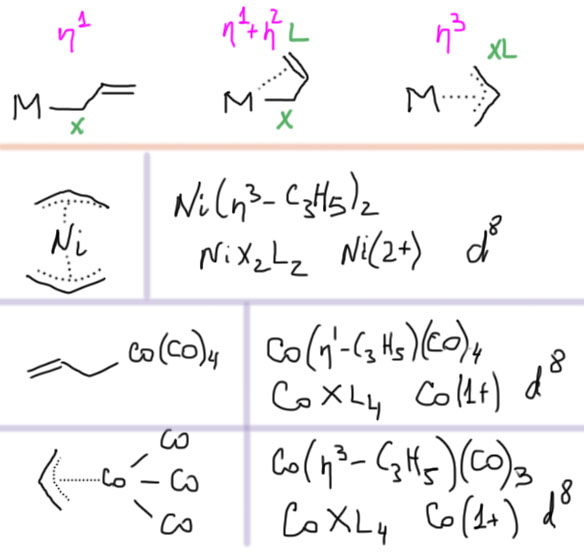
Аллильный остаток сильно отличается от уже рассмотренных лигандов - это не полноценная молекула, а одновалентный остаток. Вполне очевидно, что он может связываться с металлом обычной ковалентной связью, как любой простой X-лиганд. Такие комплексы существуют и их много. Двойная связь аллила может сесть на атом металла как дигапто-L-лиганд. Поскольку три атома углерода, вовлечённые в эти взаимодействия рядом, скорее мы имеем дело с единым лигандом, имеющим η3-связывание. В этом случае это четырёхэлектронный лиганд, учитываемый как X+L.
В растворах аллильные лиганды двух типов в комплексах обратимо превращаются друг в друга, что приводит к переползанию металла с одной стороны плоской аллильной системы на другую.
От аллильного лиганда один шаг до самого знаменитого лиганда металлоорганической химии - циклопентадиенила. Это шестиэлектронный η5-лиганд, который для удобства учитывают как 2L+X.

С металлами и лигандами разобрались – теперь попробуем собрать все вместе в комплекс (координационное соединение), и попробуем посчитать электроны, вовлеченные в образование таких молекул, и посмотрим, что из этого получится. Считать придется по очень строго сформулированному правилу, которое гарантирует, что будет получаться одно и то же, кто бы ни считал. Из такого счета получается знаменитое и очень полезное правило 18-электронное правило.

Чтобы посчитать валентные электроны в координационном соединении, нужно сначала записать его обобщенную формулу (структурный тип), разобрав лиганды по типам. Дальше можно воспользоваться одним из двух способов - ионным или ковалентным, - или сразу обоими для самопроверки. Оба способа должны дать один и тот же результат.
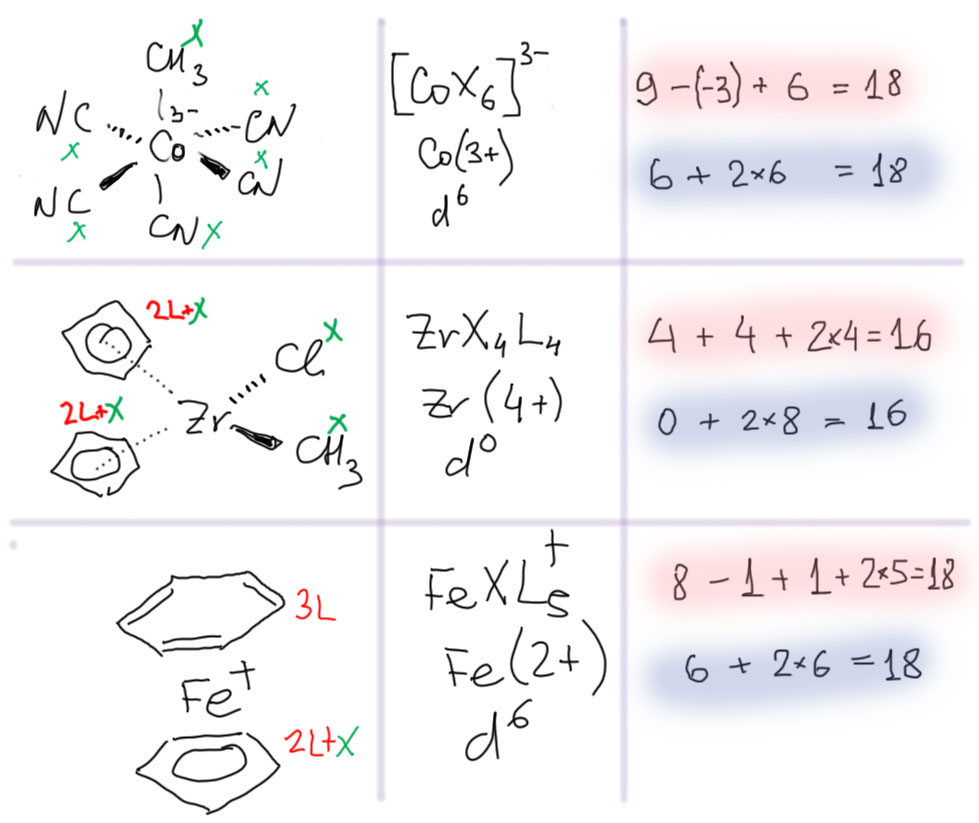

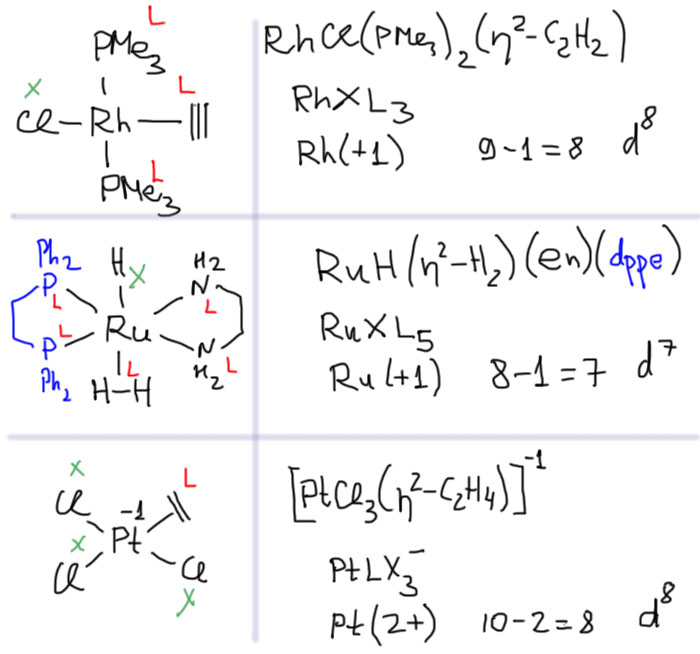
Попробуем посчитать электроны в нескольких комплексах, взятых наугад. Слева нейтральные комплексы, справа заряженные. Красным подсвечен расчет по ковалентной модели, синим – по ионной. Как видим, получается одно и то же. 18-электронные комплексы. встречаются чаще 16-электронных.
16-электронные комплексы среди этих примеров это, с одной стороны, комплекс раннего переходного металла. С другой стороны, это плоско-квадратный комплекс позднего переходного металла.

Одно из важнейших следствий счета электронов – представление о том, что 18-электронные комплексы не только встречаются чаще всех остальных, но и обладают интересной особенностью – координационной насыщенностью, то есть принимают максимально возможное число лигандов для данного валентного состояния металла. Если есть координационная насыщенность, то есть и координационная ненасыщенность – состояние, подразумевающее возможность изменения количества лигандов на металле, а это совершенно необходимо для того, чтобы комплекс металла мог участвовать в химической реакции, связанной с изменением лиганда. А это именно то, что нам нужно для того, чтобы стало возможным то, о чем мы будем говорить в этом курсе.
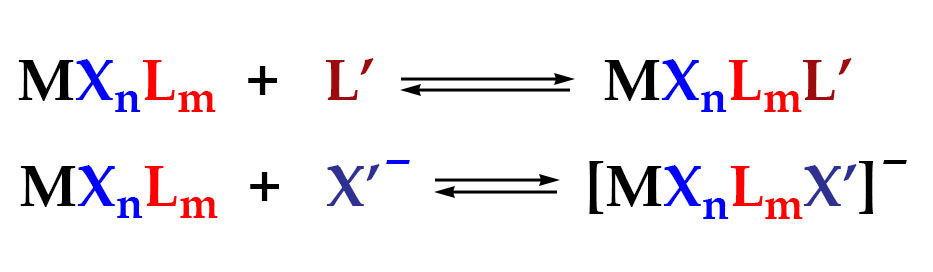
Координационная ненасыщенность проявляется в способности комплекса присоединять еще один лиганд любого типа. Равновесия присоединения-отщепления лиганда (лигандного обмена) играют особую роль в химии переходных металлов.
Комплексы с 18 электронами являются координационно насыщенными
Это очень полезное правило, которое позволяет оценивать емкость координационной сферы металла в реакциях, когда лиганды приходят, изменяются и уходят - нужно следить за тем, чтобы количество лигандов в координационной сфере не превышало количества, создающего 18-электронный счет. Комплексы с меньшим числом электронов принципиально могут вступать в реакции, сопровождающиеся увеличением счета электронов до 18 даже в том случае, когда исходные комплексы проявляют свойтсва координационно насыщенных.
В связях металлов с лигандами часто присутствует и дополнительное взаимодействие, называемое по-английски back donation, за счет взаимодействия заполненных орбиталей металла dπ-типа с вакантными орбиталями лиганда, подходящими по симметрии, а это обычно разрыхляющие π-орбитали. Это взаимодействие играет совершенно колоссальную роль как в структурах комплексов, так и в их реакционной способности. В каком-то, а точнее, во вполне определенном смысле это взаимодействие является ключевой причиной того, что лиганды в координационной сфере металла приобретают новую реакционную способность, и вступают в разнообразные реакции. Можно даже сказать, что если бы не было этого эффекта, не было бы и этого курса лекций, потому что не о чем было бы рассказывать.
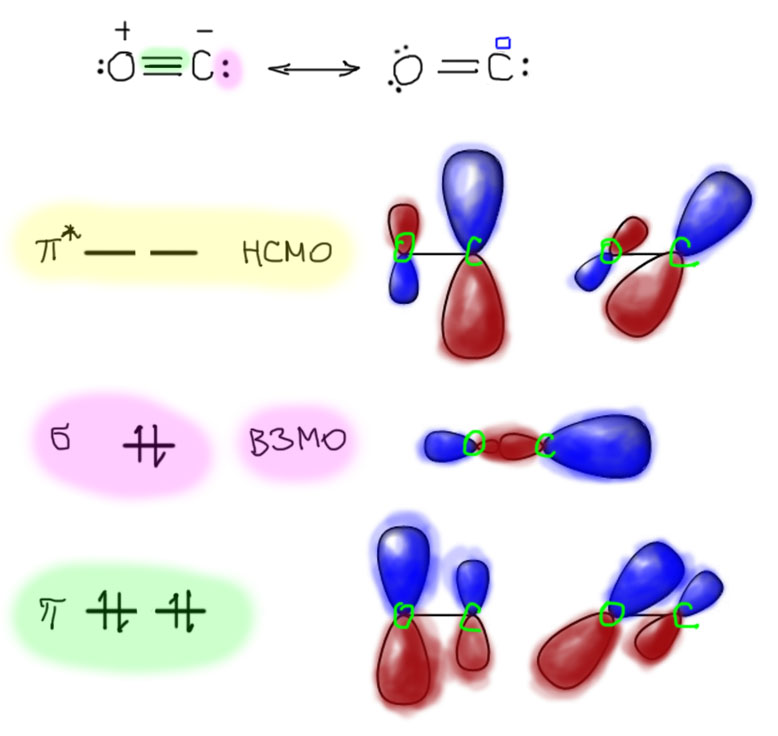
Оксид углерода или просто карбонил - один из важнейших лигандов в химии переходных металлов. В большинстве комплексов это сигма-донорный лиганд, связанный с металлом за счет донорно-акцепторной связи, образуемой за счет ВЗМО молекулы CO, которую можно легко отождествить с неподеленной парой на атоме углерода. Стандартная форма карбонильного лиганда не сильно отличается от свободной молекулы CO c sp-гибридными атомами и тройной связью.
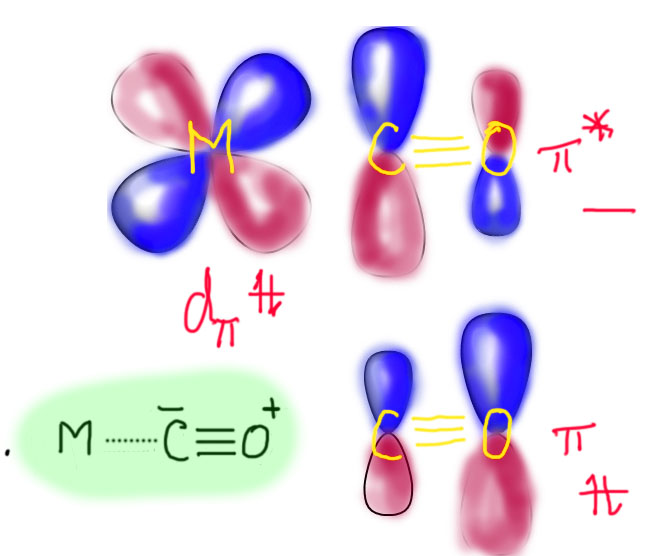
Кроме координационной сигма-связи в карбонилах металлов часто возникает еще один тип связывания, который называют back-donation. Во многих карбонильных комплексах, особенно низковалентных поздних переходных металлов возникает дополнительное связывание за счет вакантной разрыхляющей орбитали оксида углерода и заполненной d-орбитали металла. В этом взаимодействии смещение электронной плотности происходит от металла к лиганду, а следовательно лиганд ведет себя как кислота Льюиса, а металл как основание. Поскольку back-donation это π-связь практически всегда (бывает ещё дельта-связь, например, в том же дибензолхроме) лиганд, способный на back-donation, часто называют π-кислотой (металл, способный на back-donation это π-основание).
Back-donation, как правило, действует не как полноценная химическая связь, а скорее как электронный эффект, вызывающий смещение электронной плотности от металла на лиганд. Это смещение может быть и очень слабым, но может проявляться очень сильно в значительном упрочнении связи металл-лиганд, и одновременном ослаблении (разрыхлении) связей в самом лиганде. Этот эффект поэтому играет чрезвычайно важную роль в влиянии металла на свойства и реакционную способность лиганда. Молекула, войдя в координационную сферу металла, приобретает высокую реакционную способность в различных реакциях, не свойственную этой молекуле в свободном состоянии. Это явление называют активацией.
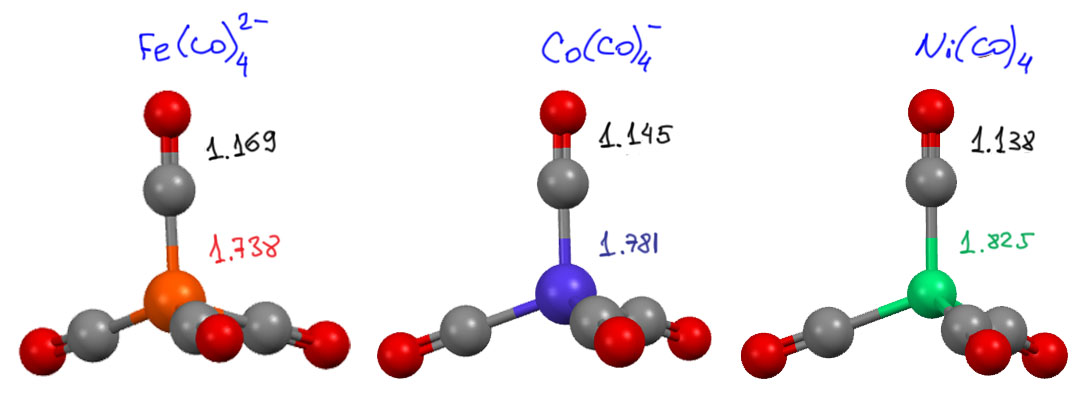
У низковалентных комплексов ранних переходных металлов back-donation может играть главную роль, обусловливая основной вклад в связывание лигандов с металлами. Этот эффект приводит к парадоксальным и не очень легко оцениваемым изменениям - изменяется степень окисления металла, счёт d-электронов, структура лиганда. Совершенно особая химия ранних переходных металлов, особенно из самого начала блока d-элементов, во многом связана с этим эфектом.
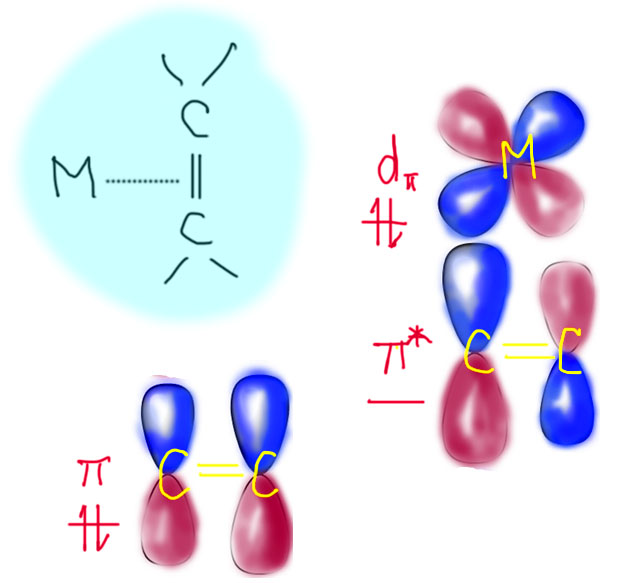
Другой важный пример back donation дает связь переходного металла с олефином. В этом случае для связи используется разрыхляющая орбиталь двойной связи. Перенос электронной плотности с металла на эту орбиталь закономерно приводит к ослаблению двойной связи, и этот эффект часто используют в реакциях олефинов с комплексами переходных металлов.