
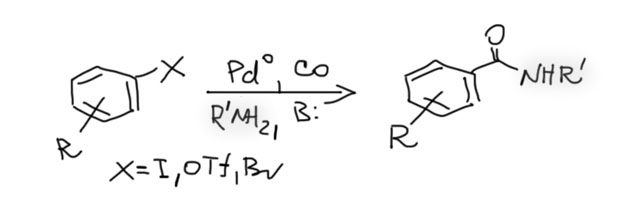
Реакции с участием оксида и диоксида углерода
Углерод как элемент тверд и поэтому малопригоден для нормальных реакций, так как образует в твердой фазе бесконечные двухмерные или трехмерные цепочки прочно связанных друг с другом атомов, которые нужно сначала как-то контролируемо разрушать. Это требует огромных затрат энергии, и, соответственно, условий, трудносовместимых с нормальными реакциями синтеза. Для того, чтобы участвовать в хороших, то есть разнообразных и синтетически продуктивных реакциях, углерод должен быть превращен во что-то простое, но более доступное в виде отдельных молекул, на которые можно культурно воздействовать реагентами и катализаторами. Самые простые молекулярные представители элемента углерода – оксид и диоксид углерода, а также метан. Эти три соединения составляют то, что часто называют C1-химией, имея в виду, что преобразование одноуглеродных молекул в более сложные – достойная задача органического синтеза, позволяющая воспроизводить все многообразие органических молекул из очень простой и практически неограниченной сырьевой базы. Мы отлично знаем, что с этой задачей превосходно справляются бактерии и растения, и нам как-то неудобно считать себя синтетиками низшего порядка по сравнению с ними, хотя это действительно так и есть, и было бы мудрее смириться. Про метан мы поговорим отдельно, а здесь займемся оксидами углерода.
Из двух обычных оксидов углерода, монооксид CO обладает невероятно высоким авторитетом в химии переходных металлов. Оксид углерода или просто карбонил – один из самых важных и наиболее подробно исследованных лигандов, а реакции этой простой молекулы многочисленны и разнообразны. Это само по себе отлично, но есть одна проблема – оксид углерода не является непосредственно доступным производным углерода – он, к счастью, не встречается на планете Земля в ощутимых количествах, его нужно делать из угля (в промышленности), или муравьиной кислоты (в лаборатории). Это вещество весьма токсично, при этом оно не обладает ни вкусом, ни запахом, что делает его чрезвычайно опасным – люди незаметно травятся оксидом углерода насмерть. В лабораториях и промышленных помещениях, где работают с оксидом углерода, используют всякие способы распознать опасную концентрацию, от автоматических датчиков до бумажек, пропитанных солями палладия. Дурная слава незаметного убийцы, тянущаяся за этим веществом, лишает многих всякого желания встречаться с ним в работе и жизни. В лабораторных условиях существует множество способов генерировать CO прямо в реакционной смеси из более безобидных или, по крайней мере, осязаемых веществ, но дискомфорт все равно остается.
Диоксид углерода в смысле доступности и опасности отличается от CO радикально. Колоссальные количества этого вещества находятся в атмосфере и ежесекундно вырабатываются при сжигании топлива и жизнедеятельности живых организмов. Проблема глобального потепления как раз и связана с повышением концентрации диоксида углерода в атмосфере, и извлекать его оттуда было бы очень выгодно. На самом деле, это было бы так, только если бы это удавалось делать в совершенно циклопических масштабах и только в том случае, если бы эта деятельность происходила бы за счет энергии возобновляемых источников. Поскольку ни о том, ни о другом речь не идет, нам остается только удовлетвориться обычной отговоркой, что с чего-то нужно начинать. Диоксид углерода нельзя назвать совсем безопасным газом, и лишиться жизни с его помощью тоже можно, но все же и концентрация для этого потребуется на порядки более высокая, и накопление реально опасных концентраций этого газа требует особых условий – замкнутых помещений, и почти полного отсутствия газообмена. Недавняя трагедия в бассейне, куда для понта вывалили мешок сухого льда, показывает, как легко организовать такую бойню с помощью с виду безобидного прикольного материала. Химики хорошо знают, что каждые 44 грамма твердой углекислоты выделяют в воздух 22.4 литра газообразной, и если мы сдуру быстро испарим всего 4.4 кг получим два с лишним кубометра. Откачать не успевают – CO2 растворяется в крови и изменяет кинетику переноса кислорода гемоглобином. Казус известных пещер вокруг супервулкана Флегрейских полей в окрестностях Неаполя, где плохо себя чувствуют маленькие собаки, а люди даже не замечают проблем, показывает еще одно спасительное свойство этого тяжелого газа – он скапливается в опасных концентрациях только снизу. В общем, если не делать глупостей – не реагент – мечта! Еще и приплачивать будут, если вы его куда-нибудь засунете. Увы, это оказывается настолько непросто, что реальных успехов в химии диоксида углерода пока что кот наплакал. Почему все так печально, посмотрим, когда доберемся до этой интересной, но немного импотентной молекулы.
Начнем с оксида углерода.
Оксид углерода - отличный реагент в синтезе не только лабораторном, но и промышленном. Реакции CO - одни из первых промышленных каталитических реакций. В лаборатории используют или баллоны с CO (кто не боится и имеет очень хорошие тяги) или разложение муравьиной кислоты концентрированной серной.
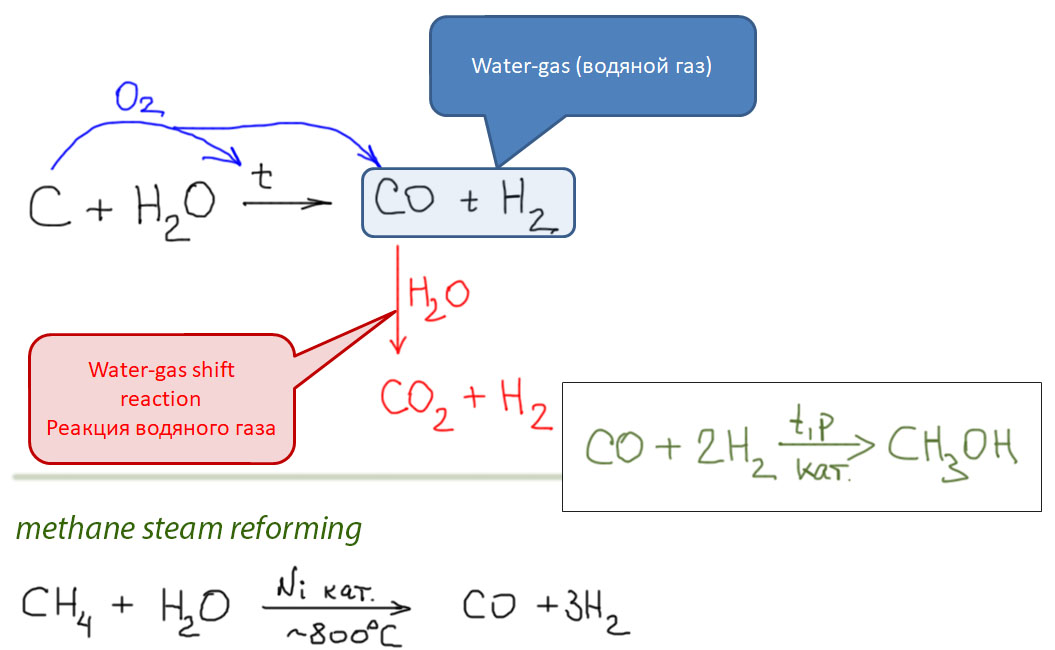
В промышленности очень часто используют не чистый CO, а его смеси с водородом в соотношениях 1:1 и 1:2. Основной источник оксида углерода для большого синтеза - конверсия угля, взаимодействие угля с водяным паром при очень высокой температуре, которую поддерживают чередованием водяного пара с воздухом. При этом образуется смесь CO и водорода, исторически называемая водяным газом. Соотношение CO:H2 регулируют с помощью так называемой реакции водяного газа (water-gas shift reaction) - для разных реакций требуются смеси от 1:1 до 1:2, такие смеси часто называют синтез-газом.
Есть еще и вторая реакция, которая дает смесь CO и водорода, но с большим содержанием водорода - паровой риформинг метана, гатерогенная каталитическая реакция. Но эту реакцию применяют в основном для производства водорода, а СО из газовой смеси удаляют с помощью реакции водяного газа.
Самая важная реакция с синтез-газом c соотношением CO:H2 2:1 - синтез метанола, а метанол далее перерабатывают в формальдегид, уксусную кислоту и многие другие важные соединения.
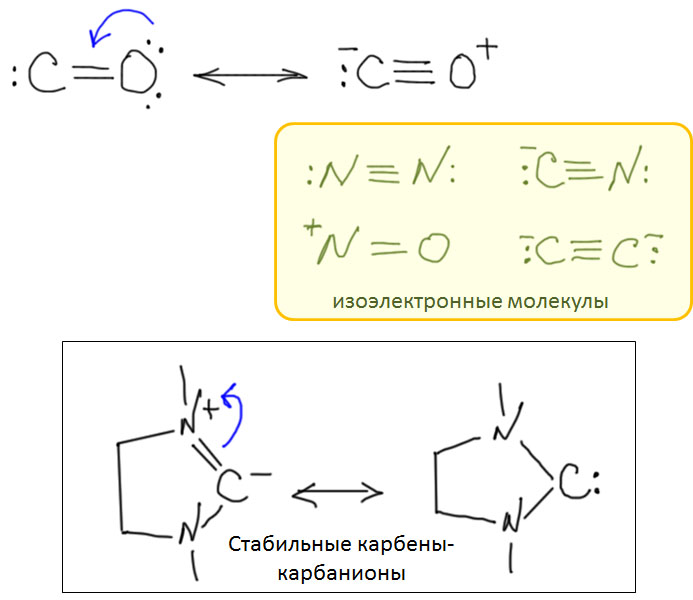
CO - маленькая, но очень интересная молекула. В свободном состоянии (не в виде лиганда в комплексах) эта молекула ближе к структуре с тройной связью и зарядами на атомах - можно даже сказать, что это карбанион (причем илидного типа, так как рядом положительно заряженный атом кислорода), только не забывать, что соответствующая ему сопряженная кислота - это формильный катион, очень сильная CH-кислота, а следовательно, CO - это очень слабое основание Бренстеда. Если рассматривать CO как основание Льюиса, то мы хорошо видим, что это именно мягкое основание Льюиса - мягкие основания Льюиса всегда являются слабыми основаниями Бренстеда. Вторая граничная структура имеет карбеновый характер. Эта структура дает небольшой вклад в структуру свободного CO, но играет очень большую роль в комплексах.
Очень интересно и поучительно рассматривать CO в ряду изоэлектронных частиц, в которых проявляются оба структурных типа, более или менее, в зависимости от природы второго атома - от чисто анионного (карбанионного) типа, как в ацетиленид-ионе, до чисто карбенового, как в нитрозоний-катионе (точнее, нитренового, но нитрен - это это просто аза-аналог карбена с тем же сочетанием пустой и заполненной орбитали на одном атоме). Посредине - цианид, он же изоцианид-ион, в структуре которого карбеновая и карбанионная формы представлены одинаково значительно. От цианид-иона аналогия, как мы уже знаем, приведет нас к гетероциклическим карбенам, также проявляющим дуализм карбанион-карбен.
Лиганд CO официально называется карбонилом. В комплексах с переходными металлами CO (которые также называются карбонилами) образует как обычную координационную связь (σ-связь) за счет пары электронов на углероде, так и вторую связь (π-связь) за счет обратного донорного эффекта (back-donation). Второй эффект зависит от возможностей металла и свойств других лигандов. CO - один из самых сильных π-акцепторов среди лигандов. Если вклад π-связи невелик, в структуре лиганда преобладает троесвязанная форма, если велик - порядок связи снижается в пределе до двух, между металлом и карбонилом в этом случае можно рисовать двойную связь. Такая структура немного напоминает же знакомый нам винилиденовый карбен в комплексах. Карбонильный лиганд очень удобен тем, что позволяет легко диагностировать вклад back-donation по ИК-спектру, так как частота валентного колебания связи углерод-кислород сильно и очень характерно зависит от ее порядка, снижаясь при уменьшении порядка.
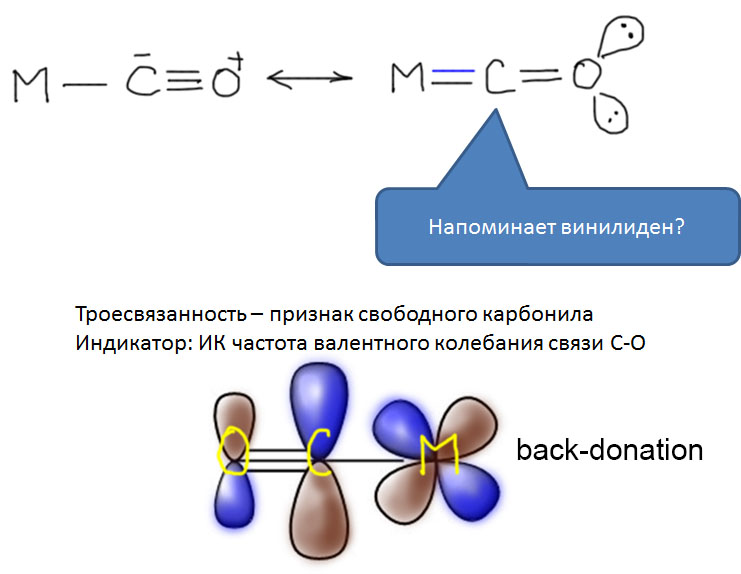
В отличие от настоящих карбенов, карбонил принимает back-donation на разрыхляющую орбиталь π-связи с кислородом, в основном находящуюся (имеющую больший вес) на атоме кислорода - то есть электроны с металла фактически поступают на электроотрицательный кислород, создавая там отрицательный заряд. Это очень выгодно, поэтому карбонил так хорошо стабилизирует низкие степени окисления металлов, причём мы должны понимать, что формальная степень окисления металла в таких комплексах может нас обманывать. Если совсем уж осмелеть в аналогиях, то можно увидеть что в комплексах с низковалентными и ранними переходными металлами карбонил ведёт себя как карбен Шрока. Но - будьте осторожны, здесь нужно сохранять некоторую степень благоразумия, так как в координационной химии монодентатный карбонил всегда рассматривают как L-лиганд.
Характерным примером такого поведения карбонильного лиганда являются анионные карбонилы металлов, известные для всех переходных металлов кроме 10 и 11, а также 3 групп (J. E. Ellis Organometallics 2003, 22, 3322). По свойствам эти комплексы всегда являются сильными восстановителями и довольно часто - сильными нуклеофилами. Формально, при учёте карбонила как L-лиганда в этих комплексах металл имеет максимальное для своей группы число d-электронов, совсем нехарактерное в первую очередь для ранних переходных металлов.
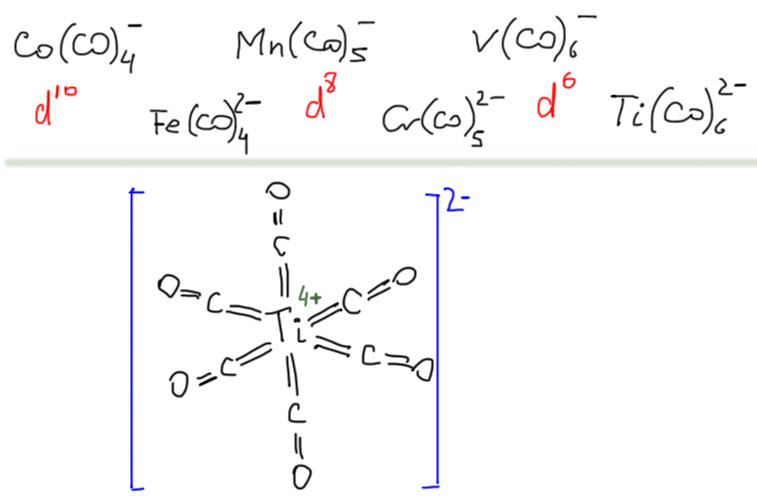
При этом, если мы учтем вклад back-donation в полной степени, то получим, что, например, у комплекса металла 4 группы шесть карбонилов в пределе снимают всю плотность с металла и вместо формальной степени окисления 2-, очень странной для раннего переходного металла, получим обычную степень окисления 4+, то есть обычную для этой части d-блока. В принципе, это очередное повторение фокуса с фактическим изменением степени окисления при максимальном вкладе back-donation, которое мы уже видели и в металлациклопропанах, и в карбенах Шрока.

Итак, CO легко входит в координационную сферу переходных металлов, как правило, образуя карбонильные комплексы с моногапто-связыванием молекулы CO через углерод. Другие типы связывания – и дигапто-, и мостиковые – тоже бывают, но играют относитеьно небольшую роль в реакциях с участием органических соединений. Посмотрим, что происходит дальше, уже в координационной сфере.
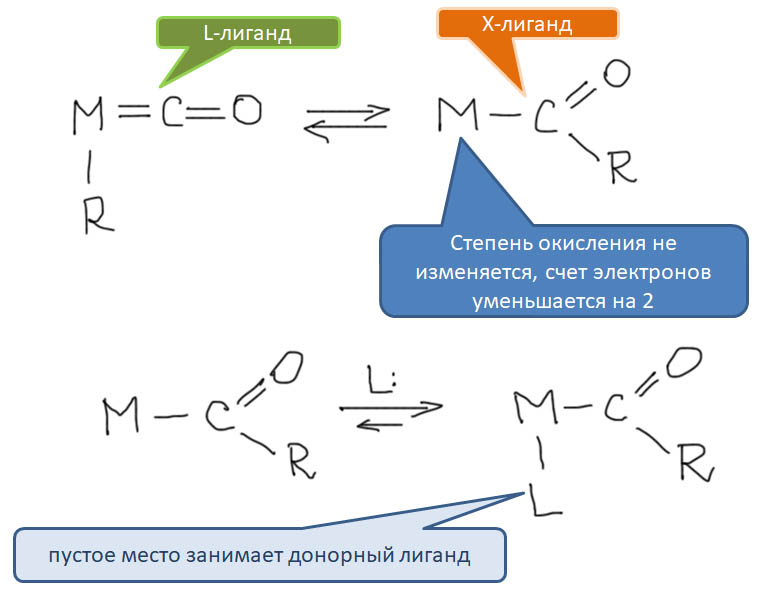
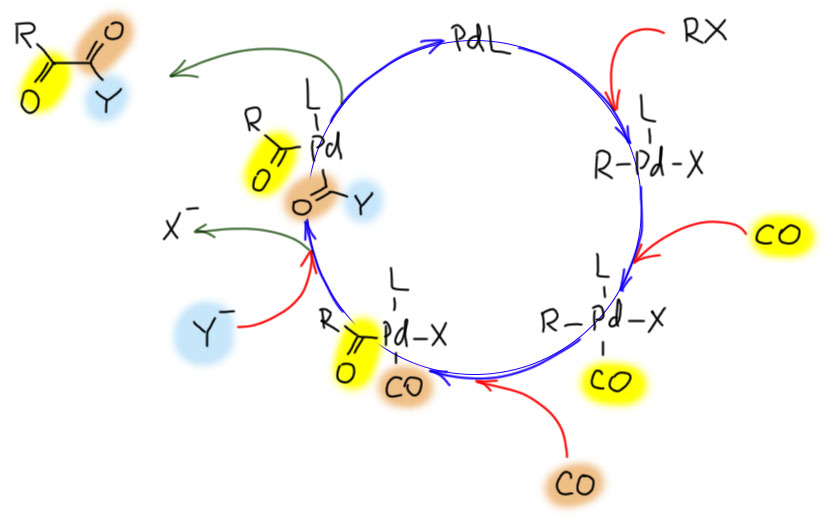
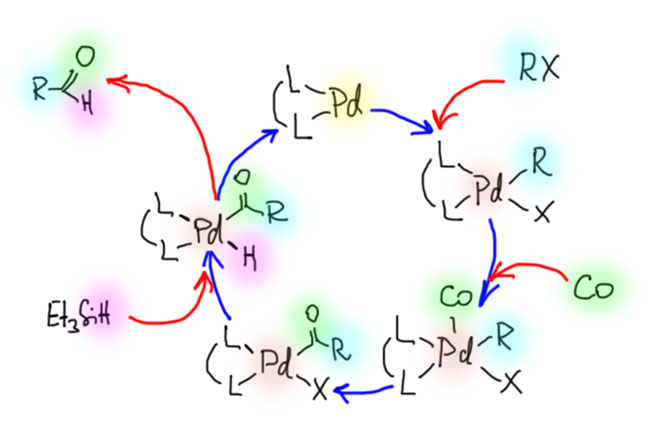
Аналогия карбонильного лиганда и карбенов распространяется и на реакции. Основная реакция в координационной сфере металла - фактически та же самая, что часто происходит с карбеновыми лигандами - миграционное внедрение σ-связанного X-лиганда на углеродный атом карбонила (карбена), непосредственно связанный с металлом. Эта реакция обратима, и смещение равновесия в сторону продукта внедрения обеспечивается связыванием донорного лиганда из реакционной смеси (фосфина, еще одной молекулы CO или хотя бы донорного растворителя). Степень окисления металла не изменяется, как и в других реакциях миграционного внедрения. Продукт миграционного внедрения - σ-ацильный комплекс.
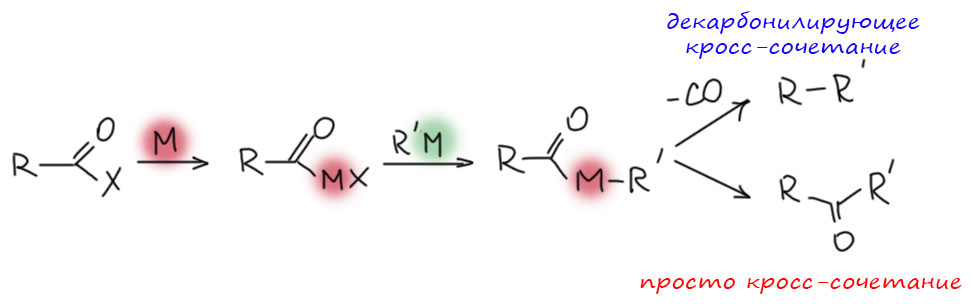
Обратимость реакции миграционного внедрения может быть использована довольно неожиданным образом. Можно получить σ-ацильный комплекс по-другому, например, окислительным присоединением к ацил хлоридам или другим производным карбоновых кислот. Такой ацильный комплекс может или участвовать в обычном кросс-сочетании с образованием кетонов, либо потерять CO и направиться по пути декарбонилирующего кросс-сочетания или реакции Мидзороки-Хека.
Один из фокусов, которые можно встретить в этой химии - реакция Мидзороки-Хека без участия оснований. Такую реакцию можно сделать, если взять в качестве электрофила ангидриды или амиды ароматических кислот - уходящая группа в этом случае играет роль основания, а выход этой группы из координационной сферы обеспечивается лигандным обменом с галогенидами..
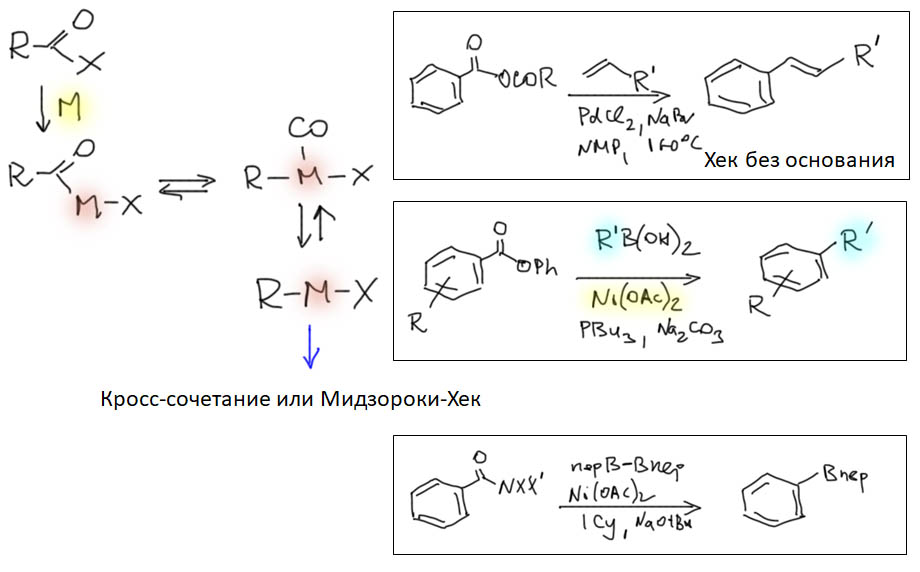
Кроме обращения миграционного внедрения, ведущего к потере карбонила, ацильные комплексы могут вступать и в другие реакции, при которых карбонил сохраняется. Есть три основных пути, встречающихся в огромном количестве процессов.
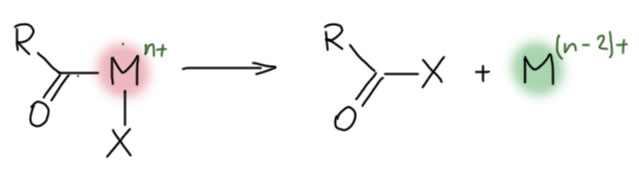
Восстановительное элиминирование происходит, если на металле есть ещё один окисляемый лиганд, а металл имеет не самую низкую степень окисления. Эта реакция прежде всего встречается в карбонилирующем кросс-сочетании и родственных процессах. Металл изменяет степень окисления на -2.
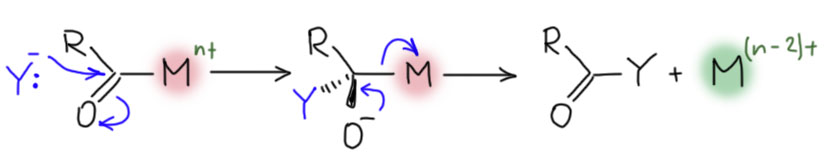
Другая очень распространённая реакция - расщепление ацила нуклеофилом. Эта реакция чертовски похожа на нуклеофильное замещение в карбонильных соединениях - также идёт через тетраэдрический аддукт. Металл, как и в восстановительном элиминировании, уменьшает степень окисления на 2.
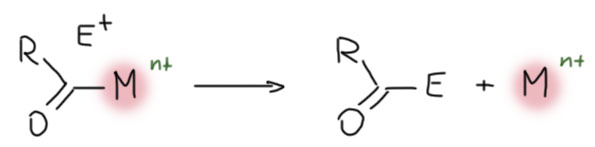
Третья реакция, немного более редкая, - расщепление ацильного комплекса электрофилом. Чаще всего в роли электрофила выступает протон (протонная кислота), тогда реакцию называют протолизом. Металл в этой реакции не изменяет степени окисления.

Теперь обратим наше внимание на применение карбонилов и оксида углерода в синтезе. Начнем с очень симпатичной химии, ключевую роль в которой играют высоконуклеофильные карбонилаты металлов. Эта химия использует супернуклеофильность анионных карбонилов, которые очень охотно вступают в SN2-замещение с типичными для этой реакции субстратами. Эта химия начиналась со стехиометрических реакций карбонилатов железа, но развитие получила в каталитических реакциях с участием легкодоступного карбонилата кобальта. Именно с исследования этой химии начал свой невероятный путь в химии – от Нобеля к нобелю – Ричард Хек. Хотя эти реакции ограничены высокореакционноспособными субстратами, пригодными для SN2-замещения, то есть, в основном, с бензилгалогенидами и некоторыми другими соединениями, но в ряде случаев эта химия эффективна и полезна.
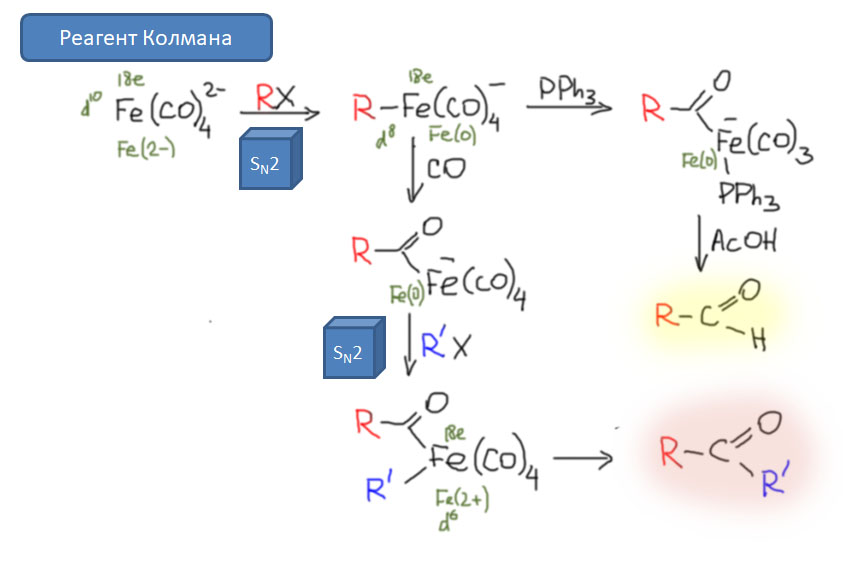
Первый удачный пример применения карбонильных комплексов в синтезе органических карбонильных соединений - использование анионного карбонилата железа (реагента Колмана) в работах Джеймса Колмана, одного из первопроходцев использования переходных металлов в синтезе. Это высоконуклеофильное производное легко реагирует в SN2-замещении, а образующиеся алкильные комплексы претерпевают миграционное внедрение с образованием ацильных комплексов. Необратимость внедерения обеспечивается или добавлением донорного лиганда или проведением реакции в атмосфере CO.
Так как исходный карбонилат - дианион, - возможно и второе замещение уже в ацильном комплексе, сохраняющем достаточную нуклеофильность. Протолиз моноалкильного комплекса дает альдегиды, а из дизамещенных комплексов уже Fe(2+) продукт, кетон, образуется спонтанным восстановительным элиминированием. Эта реакция в начале 1970-х произвела огромное впечатление на органиков, так как показала исключительный потенциал комплексов переходных металлов в органическом синтезе. Но широкому применению препятствовало то, что эти реакции были стехиометрическими и требовали работы с большими количествами токсичных и непростых в работе карбонилов.
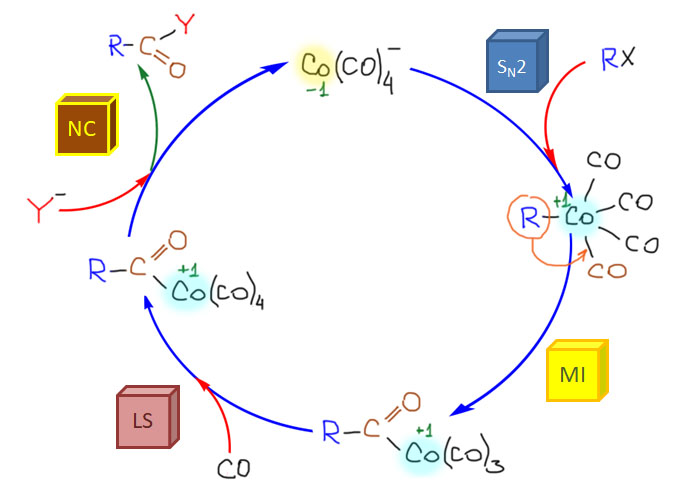
Каталитической эту реакцию удается сделать с помощью ближайшего аналога тетракарбонилферрата. К этой реакции пришёл в 1963 Ричард Хек в результате исследования механизма гидроформилирования в самом начале своей карьеры. Нуклеофильность тетракарбонилкобальтата тоже довольно велика, а реакцию удается продвинуть вперед к ацильному комплексу просто используя избыток CO, а не дополнительный лиганд типа фосфина.
Расщепление ацильного комплекса нуклеофилами дает карбоновые кислоты или сложные эфиры, и возвращает тетракарбонилкобальтат-анион в цикл. Реакция хорошо идет с субстратами, активными в реакциях SN2, в первую очередь, с бензильными галогенидами и α-галогенпроизводными карбоновых кислот.
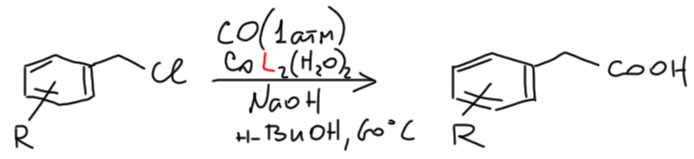
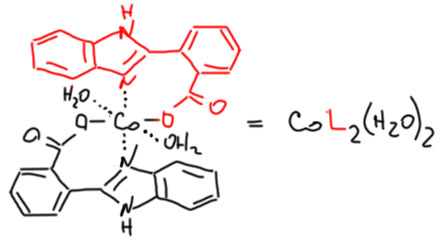
Современные версии этой реакции используют не неудобный карбонил кобальта или карбонилаты, а несложные комплексы кобальта, которые в условиях реакции восстанавливаются до карбонилатов. Одна из последних работ в этой области, описанная Ли и сотр. из Северо-Западного университета, но не того, который близ Чикаго, а того, который в китайском городе Сиань, использут комплекс с хелатором, производным бензимидазола, достигает цели в мягких условиях.
Эпоксиды легко раскрываются нуклеофилами - мы хорошо знаем эту реакцию ещё с 3 курса. Нуклеофильный тетракарбонилкобальтат не исключение - и в этом случае в результате внутримолекулярного расщепления ацильного комплекса алкоксидом может получиться 4-членный лактон. Такие лактоны вызывают огромный интерес, прежде всего как мономеры для получения биоразлагаемых полимеров полигидроксибутиратов (ПГБ).
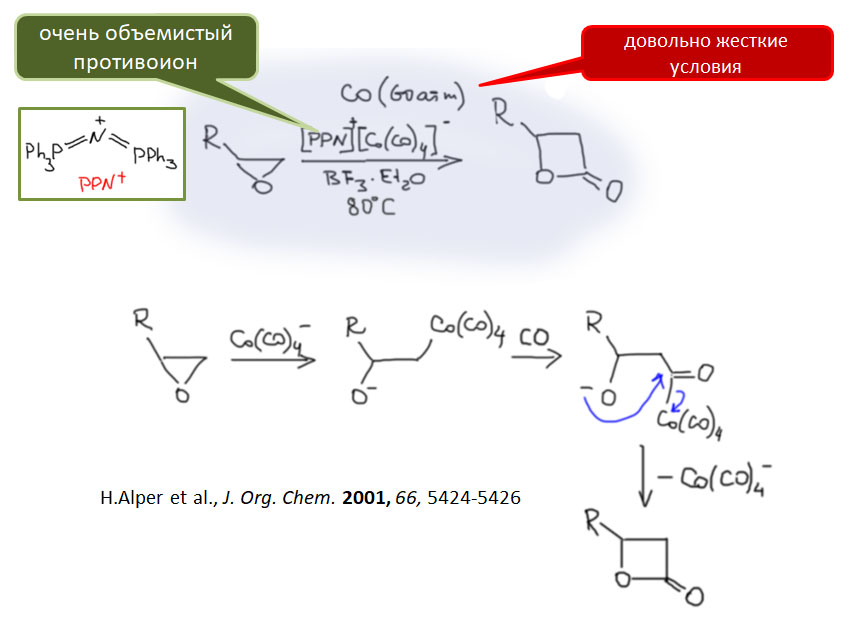
Эту реакцию открыл ещё в 1963 году Хек в стехиометрическом варианте, но сделать ее каталитической долго не получалось. Проблема в некоторой несовместимости реагентов - для раскрытия кольца с приличной скоростью требуется содействие кислотой Льюиса, но она же снижает скорость расщепления ацильного комплекса. Впервые решить эту задачу смог канадский исследователь Ховард Алпер, очень много занимавшийся самыми ранними реакциями карбонилирования. Решение нашлось в применении специфического, очень объемистого противоиона PPN и довольно жестких условий реакции. После Алпера этой реакцией занимались и другие, пытаясь как-то избавиться от этого катиона - он не очень сложен и дорог, но демонстративно неэкономичен и в смысле "экономии атомов", и в более приземлённом смысле расходов на проведение реакции и выделение продуктов.
Но гораздо более мощные методы используют не нуклеофильное замещение, а окислительно присоединение, и здесь на авансцену выходят хорошо нам знакомые металлы 10 группы – никель и палладий, а также два благородных родственника кобальта родий и иридий.
В самом общем виде каталитический цикл карбонилирования включает окислительное присоединение органического электрофила, связывание CO, миграционное внедрение и расщепление ацильного комплекса нуклеофилом - гидроксидом, алкоксидом, амином, и т.п. с образованием карбоновых кислот и их производных. Термин нуклеокарбонилирование как раз и подчеркивает эту особенность - в молекулу входит группа, состоящая из карбонила и нуклеофила, и в конкретных случаях применяют более точные термины: гидрокси-, алкокси-, амино- и т.п. -карбонилирование. При этом как раз сами карбоновые кислоты этим способом получают редко, так как гидроксид-ион в реакционной смеси вызывает назойливую побочную реакцию превращения CO в карбонат (water-gas shift reaction) и образования гидридных комплексов, расщепляющих исходные галогенпроизводные. Метод чрезвычайно популярен в синтезе ароматических кислот и их производных.
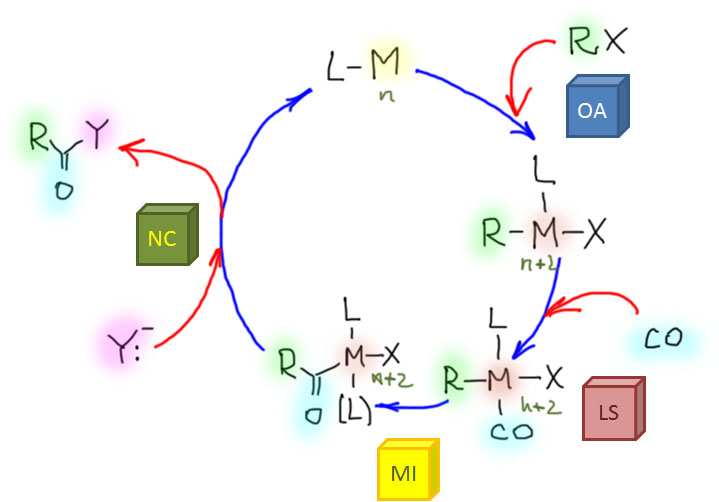
Проблема этой и аналогичных реакций в том, что оксид углерода - настолько хороший лиганд, конкурентно связываемый с металлом даже в присутствии других лигандов, вытесняемых из координационной сферы, что контролировать эту реакцию с помощью всяких специальных лигандов довольно трудно. Поэтому, в этой реакции круг субстратов (электрофилов) довольно узок и очень далек от того головокружительного многоообразия, которое мы видим в реакциях кросс-сочетания. Тем не менее, работа по конструированию более гибких и мощных каталитических систем продолжается и иногда приносит интересные результаты.
Способность тетракарбонила никеля катализировать карбонилирование (точнее гидроксикарбонилирование) ароматических галогенпроизводных была известна почти сто лет назад, но реакции требовали чрезвычайно жёстких условий (высокая температура и давление), и была поэтому описана только в патентах. Впервые запустили эту реакцию в мягких условиях, пригодных для лаборатории, и с каталитическим количеством карбонила никеля итальянские исследователи Луиджи Кассар и Марко Фоа, обнаружившие, что в полярных растворителях в присутствии гидроксид-иона реакция происходит намного быстрее и не требует жёстких условий (L.Cassar, M.Foà J.Organomet.Chem., 1973, 51, 381).
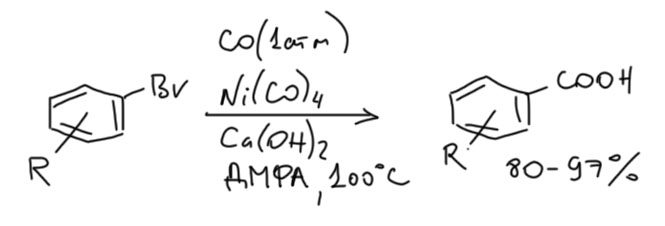
Эта работа стала основополагающей для последующего развития метода нуклеокарбонилирования. Эффект гидроксид-иона обычно связывают с образованием анионных комплексов никеля, более активных в окислительном присоединении. Загрузка катализатора, впрочем, была немаленькой (25 моль%), что при учёте выходов соответствовало не более чем 3-4 каталитическим циклам.
Надо впрочем отметить, что никель не нашёл большого примененя в карбонилировании (пока не нашёл, но никель уже столько всего за последние 10 лет нашёл, что и здесь наверняка ещё последнего слова не сказал). Это очень сильно связано с тем, что реакция карбонилирования, как и реакциях Мидзороки-Хека плохо поддаётся управлению анциллярными лигандами, а именно этот приём стоит за почти всеми великими успехами катализа последних 10-15 лет.
Карбонил никеля может быть заменён на другие комплексы. Например, Альпер и Амер показали (J. Org. Chem. 1988,53, 5147), что может быть использован цианид никеля в условиях межфазного переноса. Цианид никеля в условиях реакции превращается в анионные комплексы карбонильными лигандами.
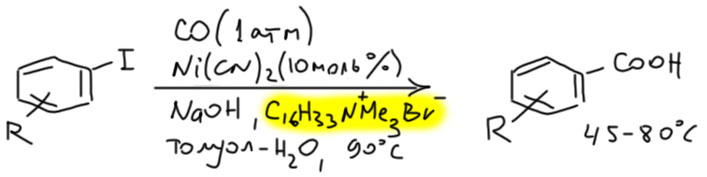
В обоих реакциях образуются соли карбоновых кислот, а сами кислоты образуются после подкисления.
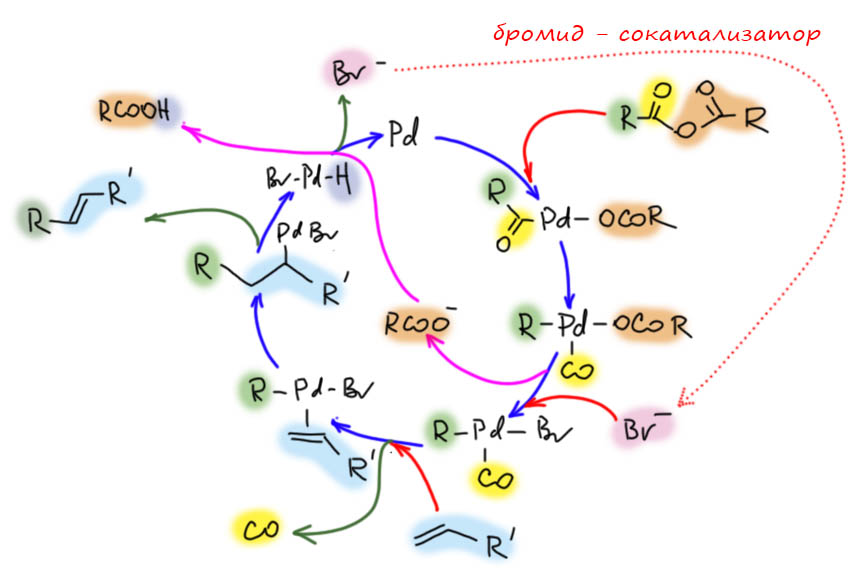
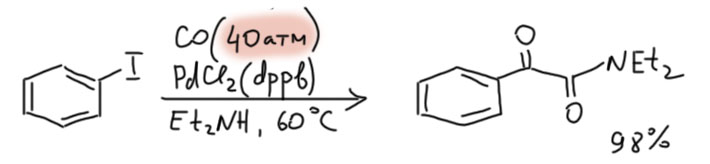
Гораздо мощнее и универсальнее в химии карбонилирования через окислительное присоединение к органическим электрофилам ведут себя катализаторы на основе комплексов палладия. Эта химия вполне аналогична химии кросс-сочетания и Мидзороки-Хека. Собственно Хек открыл и эту реакцию, описав ее в колоссальной статье 1974 года (A. Schoenberg, I. Bartoletti, R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326;). Фосфиновые комплексы палладия отлично катализируют образование сложных эфиров из иод и бром-производных бензола и алкенов. Для иодпроизводных можно ограничиться бесфосфиновым катализом и использовать просто ацетат палладия. Стоит заметить, что CO очень быстро восстанавливает соли палладия до палладиевой черни, но в присутствии иодпроизводных окислительное присоединение идёт настолько быстро, что Pd(0) не накапливается. В этом случае, очевидно, происходит такое же резервирование активного палладия через наночастицы, играющие роль resting state.
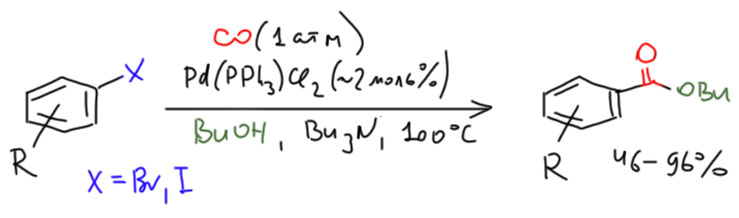
Обратим внимание, что для реакции не требуется алкоксид, но вполне работает сам спирт в присутствии не очень сильного основания, третичного амина. Хек по какой-то причине чертовски любил трибутиламин, в последующих работах, где этот протокол применялся в реальных синтезах, чаще берут триэтиламин. Спирты тоже не ограничены бутанолом - можно брать другие спирты.
Непредельные (винильные) бром и иодпроизводные превращаются в непредельные (акриловые) кислоты в таком же протоколе алкоксикарбонилирования. Реакция в основном протекает с сохранением конфигурации, хотя некоторая изомеризация происходит.
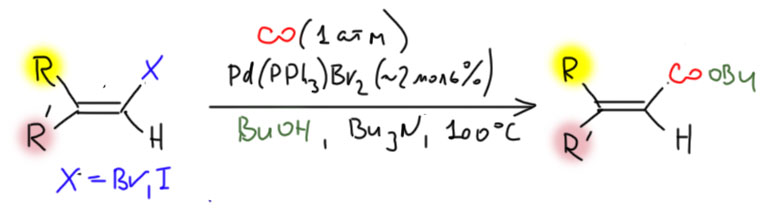
Расщепление ацильных комплексов аминами даёт амиды. Эту важнейшую реакцию также открыл Ричард Хек (A. Schoenberg, R. F. Heck J. Org. Chem. 1974, 39, 23, 3327).
В реакцию вступают первичные и вторичные амины и анилины, но не аммиак. Несмотря на то, что амины сами по себе приличные нуклеофилы способные расщепить ацильный комплекс, реакцию ведут в присутствии третичного амина. Не называйте эту реакцию, как иногда неверно делают, амидокарбонилированием или, тем более, амидированием. В реакцию вступают бром и иодпроизводные бензола и алкенов. В дальнейшем реакцию немного оптимизировали и она стала очень популярной в синтезе амидов.
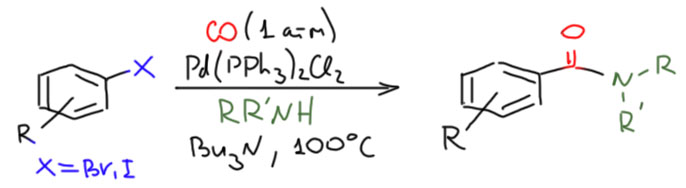
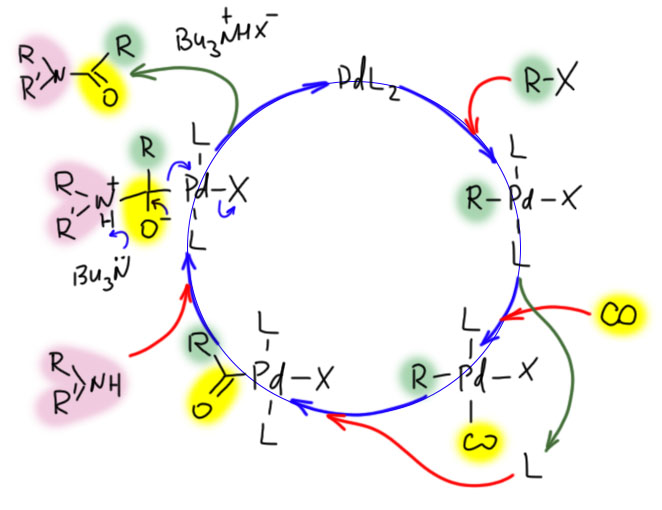
Каталитический цикл вполне типичен для нуклеокарбонилирования. Третичный амин требуется для нейтрализации кислотности, возникающей при расщеплении ацильного комплекса.
Еще раз обратим внимание на то, что анциллярные лиганды на палладии должны уметь освобождать три координационных места, чтобы впустить электрофил и молекулу CO. В этом смысле карбонилирование очень похоже на реакцию Мидзороки-Хека - в нём не работают бидентатные фосфины. Это свойство каталитического цикла очень сильно и долго ограничивало развитие метода, но в 2000-х накопление знаний о лигандах и появление новых семейств лигандов 3 поколения привело к быстрому расширению возможностей нуклеокарбонилирования.
Иногда в продукт попадает не одна, а две карбонильные группы, и образуются сложные эфиры или амиды α-кетокислот. Такой процесс называется двойным карбонилированием. Так как такие производные кетокислот - весьма полезные соединения, хороший метод их получения всячески приветствуется. Исследования показали, что механизмом такого процесса, скорее всего, является не второе миграционное внедрение CO в ацильный комплекс, а нуклеофильная атака на карбонильный лиганд с образованием второго ацила в реакции, аналогичной реакции водяного газа (water-gas shift reaction) с последующим восстановительным элиминированием продукта двойного карбонилирования.
Вполне очевидно, что такая реакция будет конкурировать с обычным нуклеокарбонилированием, но ее можно сделать основной, используя повышенное давление CO, а также подобрав правильные лиганды и условия.
Первый каталитический протокол для селективного двойного арилирования предложил Масато Танака. Протокол использует CO под значительным давлением, причём снижение давления приводит к монокарбонилированию. Также необходим хелатирующий, но гемилабильный лиганд из серии бис-дифенилфосфиноалканов, лучшим оказался dppb (T. Kobayashi, M. Tanaka, J. Organomet. Chem., 1982, 233, C64).
Новые лиганды с большим трудом нашли себе дорогу в карбонилирование. Но кропотливое изучение каталитических циклов карбонилирования открыло некоторые возможности.. Контраст с кросс-сочетанием продолжает оставаться разительным - там лигандов и протоколов уже сотни. В карбонилировании каждый новый метод сдаётся только самым упорным и опытным исследователям, и обычно никаких вариаций не подразумевает.
Очень показательна история вовлечения хлорпроизводных в нуклеокарбонилирование. Очень долго они не сдавались - появлялись только работы по отдельным, обычно гетероциклическим субстратам, а это не считается - в гетероциклах хлор настолко подвижен, что больших усилий по его замешению хоть в обычной органической химии, хоть в катализе комплексами переходными металлами не требуется. В конце концов лиганд был найден, причём всего один, причём независимо и группой Хартвига, и группой Бачуолда. Это, кажется, первый случай, когда они сошлись на практически одном решении, прийдя к нему с совершенно разных сторон. Лиганд dcypp - аналог dppp с циклогексильными группами вместо фенилов.
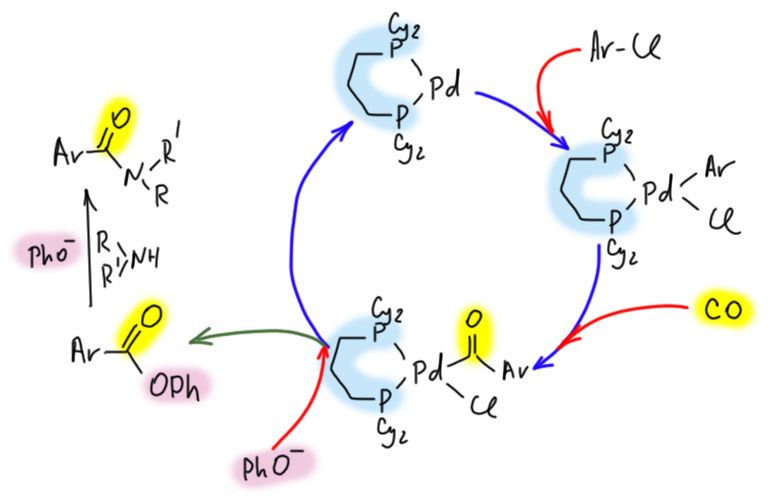
Протокол Бачуолда особенно затейлив (Buchwald S.L. Angew Chem Int Ed. 2007, 46, 8460). Он установил, что для успеха аминокарбонилирования хлорпроизводных нужен не только этот специфический лиганд, но и использование феноксида в качестве основания. Этот пример очень ясно показывает, как трудно в реакциях такого типа совместить все необходимые компоненты реакционных смесей. Бачуолд обнаружил, что в каталитическом цикле получается не амид, а сложный эфир фенола - именно феноксид расщепляет ацильный комплекс. И уже снаружи цикла происходит обычное переамидирование.
И еще довольно непонятно, как происходит связывание CO и миграционное внедрение - с хелатным лигандом просто нет места в координационной сфере. Остаётся предполагать или возможность связывания пятого лиганда - такие гипотезы часто всплывают, когда в каталитических циклах с участием Pd(2+) не удаётся обойтись четырьмя лигандами. Но мне кажется, что дело здесь просто в гемилабильности этого бидентатного фосфина - временном переходе в монодентатную форму, освобождая место для CO. Естественно, это не потому что лиганду присуще свойство учтивости и прочие сентименты, а просто потому что при повышенной температуре в реакции есть равновесие между двумя формами комплекса.
Интересно, что у Хартвига получился немного другой протокол и другие выводы о механизме каталитического цикла (J.F. Hartwig et al, JACS, 2018, 140, 7979). Но эти двое не устают спорить с 1994 года.
Тщательный подбор современных лигандов и других компонентов каталитической системы позволяет достигать впечатляющих результатов и в этой области - один за другим сдаются сложные нуклеофилы, трудносовместимые с обычными условиями палладиевого катализа.
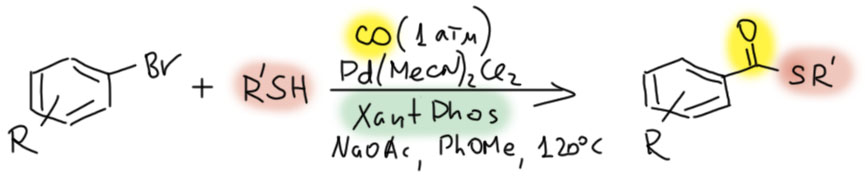
Так, датский исследователь Скрюдструп с сотр. сделали эффективный протокол для тиокарбонилирования и получения тиоэфиров (T. Skrydstrup et al J. Org. Chem. 2014, 79, 11830). Сернистые соединения всегда считались страшной отравой для палладиевых катализаторов, и это действительно так, но безусловно это относится только к гетерогенному катализу. В гомогенном катализе проблема отравления - это просто проблема защиты координационной сферы от связывания избыточного количества хорошего лиганда, и эта проблема решается подбором анциллярных лигандов для эффективного контроля координационной сферы. В этом случае опять отлично сработал XantPhos.
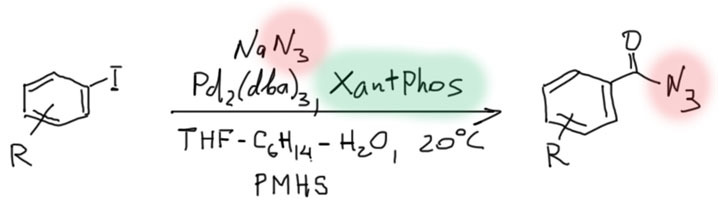
Отличный пример того, как решают проблемы каталитических процессов - разработка азидокарбонилирования. Азид - отличный нуклеофил, но задача совмещения в одной реакционной смеси азида, палладия, фосфина может показаться неразрешимой - азид с такими соседями напоминает курицу в компании волка и лисы - он реагирует и с фосфинами и с Pd(0). Кроме исходного азида нужно было умудриться сохранить и продукт реакции, ацилазид, также очень реакционноспособный и неустойчивый при повышенных температурах. Тем не менее, филигранная работа по подбору всех компонентов каталитической системы, осуществлённая Владимиром Грушиным в Каталонии привела к успеху (F. M. Miloserdov, V. V. Grushin Angew. Chem. Int. Ed. 2012, 51, 3668 –3672). Ключевым решением было использование XantPhos в качестве лиганда. Кроме собственно катализатора, важными также оказались и гетерофазная водно-органическая среда реакции, разводящая по разным фазам азид-ион и фосфин, а также добавка полимерного силана, обеспечивающая реактивацию катализатора. Протокол получился настолько эффективным, что обеспечил почти количественные выходы ацилазидов с самыми разными заместителями в ароматическом кольце.
Как получить уксусную кислоту из метанола? Формально - просто добавить карбонил. Это как раз и есть карбонилирование, которое мы сейчас обсуждаем, но - мы не умеем карбонилировать спирты, потому что нет эффективных способов осуществить окислительное присоединение по связи C-OH. Выход очевиден и мы его хорошо знаем из обычной органической химии - подменить уходящую группу. Но это хорошо в лаборатории, а здесь нужен процесс для промышленности, причём крупнотоннажной - должно работать в масштабе тысяч тонн. И, казалось бы, это тупик, но давно выяснилось, что если в реакционной смеси присутствует немного HI, то прямо в реакторе образуется иодистый метил, который уже можно карбонилировать, и дальше уже дело химической инженерии придумать, как сделать этот процесс эффективным. Начинали еще в 1950-е с катализа карбонилом кобальта (процесс BASF), но для этого требовались чудовищно жёсткие условия - сотни атмосфер и высокая температура, а каталитическая эффективность была невелика.
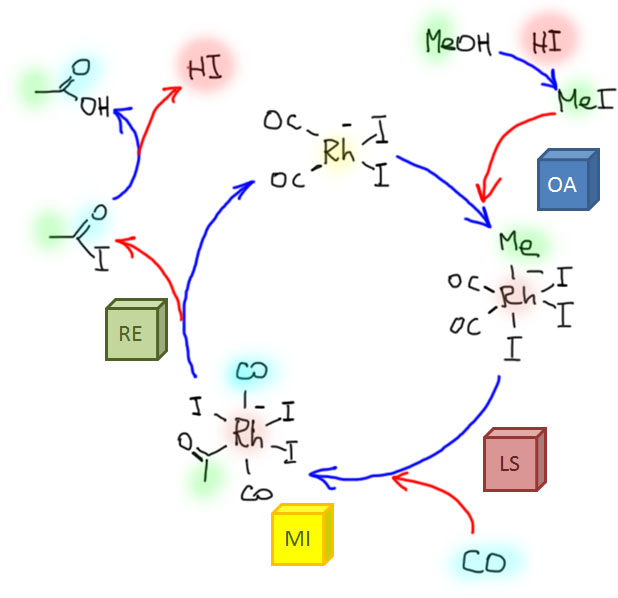
Намного более эффективен процесс, катализируемый благородным родственником кобальта родием, запатентованном компанией Монсанто в 1960-х и усовершенствованного компанией Celanese. Кроме довольно простого бесфосфинового родиевого катализатора требуется каталитическое количество HI, работающей как со-катализатор. Процесс разворачивается как типичный каталитический цикл нуклеокарбонилирования, но из-за необходимости использовать сильнокислотную среду, требующуюся для замещения гидроксильной группы в метаноле на иодид, завершением процесса не может быть нуклеофильное расщепление ацильного комплекса. Процесс завершается восстановительным элиминирование иодангидрида, быстро гидролизующегося в уксусную кислоту с регенерацией HI. Понятно что для этого требуется наличие некоторого количества воды, иначе мы получили бы метилацетат, который никому не нужен. Процесс идёт в такой сложной смеси при небольшом давлении в 30-60 атм и температуре около 200
Образующаяся уксусная кислота, вода, иодистый метил и метилацетат непрерывно отгоняются из реакционной смеси, разделяется фракционной перегонкой, и всё, кроме уксусной кислоты возвращается в реактор. Реактор функционирует с одной загрузкой очень долго, TON достигает сотен тысяч, но в конце катализатор переходит в неактивную форму, выпадающую из раствора, и тогда реактор полностью перезагружают, а родиевый комплекс регенерируют.
Метод Монсанто в 20 веке завоевал мир, став основным способом промышленного синтеза уксусной кислоты (в мире ее каждый год поизводят около 10 млн тонн, кроме карбонилирования метанола в меньших масштабах ещё окисляют ацетальдегид и, наряду с простыми кетонами и пропионовой кислотой используют катализируемое жидкофазное окисление алканов). Но в начале нового века произошло невероятное - родий, самый востребованный металл в промышленном катализе, понемногу дорожал-дорожал, да и подорожал настолько, что стал самым дорогим благородным металлом, далеко обогнав и золото, и платину. А иридий, благодаря своей исключительной редкости долгое время удерживавший звание самого дорогого металла, неожиданно отстал, и сильно.
Иридий принадлежит к той же 9 группе, что кобальт и родий, поэтому неудивительно, что он тоже способен катализировать карбонилирование метанола через метилиодид. Но долгое время считался бесперспективным и по причине дороговизны, и потому что скорость иридий-катализируемого процесса была маловата для промышленной эксплуатации.
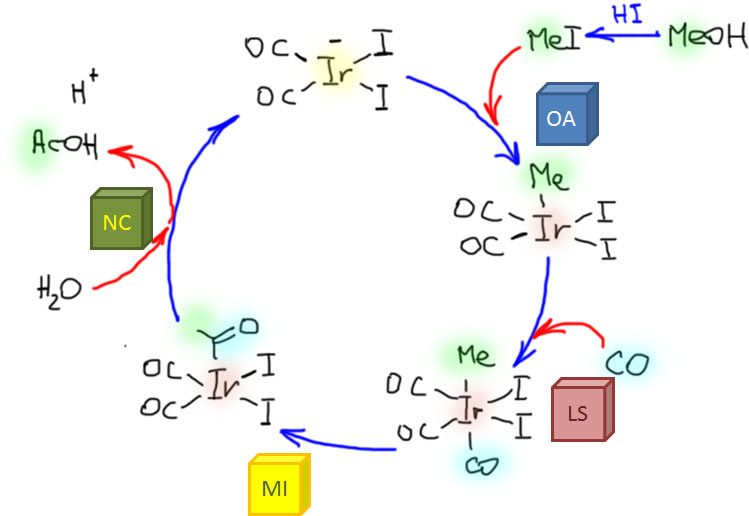
Кропотливая работа по оптимизации условий иридий-катализируемого процесса химиками компании British Petrolium, создавшей для его коммерциализации отдельную компанию Cativa, позволила преодолеть его детские болезни, и вывести его на уровень промышленной эксплуатации с экономичностью, превышающей характеристики старого процесса. Новый процесс оказался даже эффективнее процесса Monsanto-Celanese. И процесс, который безусловно доминировал более 30 лет, отправился на Свалку Истории. И теперь за каждой второй тонной банальнейшей уксусной кислоты стоит один из самых изысканных благородных металлов, иридий. И - не бойтесь, пищевые уксусы, в том числе бальзамический уксус из Модены по-прежнему делают без родия и иридия старым добрым брожением.
Иридий-катализируемый процесс очень похож на родий-катализируемый, отличаясь только деталями механизма. И уксусная кислота в нём получается именно нуклеофильным расщеплением ацильного комплекса водой. Процесс даёт намного более чистую уксусную кислоту с меньшими затратами энергии и материалов, а иридиевый катализатор намного стабильнее родиевого, и служит гораздо дольше (большие TON). Только первый завод BP Petronas Acetyls в Малайзии производит полмиллиона тонн уксусной кислоты в год.
От простого карбонилирования несложно перейти к более сложным процессам – карбонилирующим каскадам. Стадии миграционного внедрения карбонила часто встречаются в более сложных превращениях. включающих стадии кросс-сочетания, карбопалладирования, восстановительного элиминирования. Сложность получающихся продуктов ограничена только фантазией синтетика.
Карбонилирование достаточно легко соединяется в единых каталитических циклах с другими процессами, особенно часто с кросс-сочетанием, - такие процессы называют карбонилирующим кросс-сочетанием. Достаточно, но не совсем легко, потому что для эффективного управления таким каскадным процессом не хватает координационных мест у атома палладия. Поэтому такие процессы обычно довольно просты, и используют более реакционноспособные субстраты, а TON обычно невелики. Тем не менее, это довольно удобный и гибкий способ синтеза сложных кетонов.
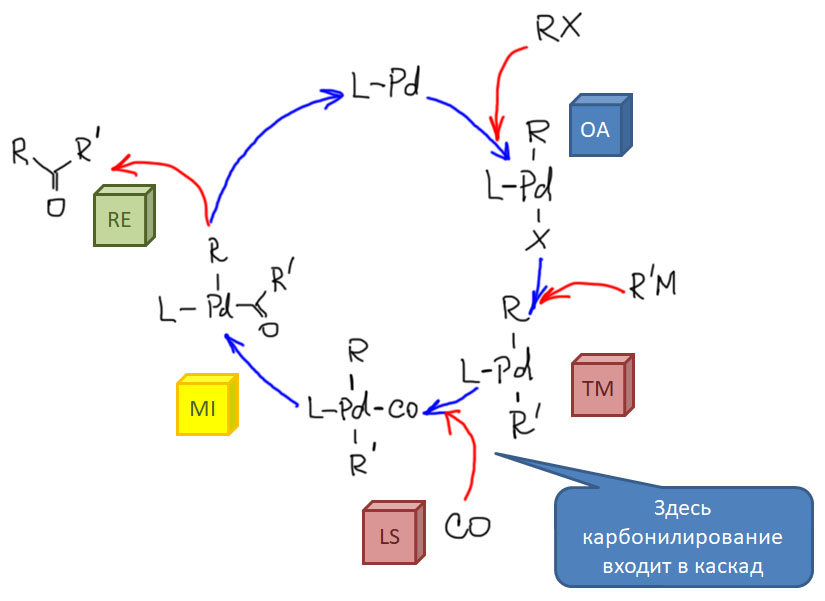
Карбонилирование обычно входит в каскад после окислительного присоединения, но или до, или после переметаллирования. В этом случае реакция практически полностью повторяет кросс-сочетание с хлорангидридами или другими производными карбоновых кислот, которое мы разобрали в самом начале лекции.
Каскадирование кросс-сочетания по Судзуки-Мияуре и карбонилирования описали сами Судзуки и Мияура с сотрудниками в 1998 году. У них получился общий метод синтеза бензофенонов с самыми разными заместителями из бром или иодпроизводных или трифлатов фенолов и бороновых кислот (T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki, N. Miyaura
J. Org. Chem. 1998, 63, 4726). Лучше всего реагируют иодпроизводные, но не только из-за лёгкости окислительного присоединения, но и потому что внедрение СО происходит легче с продуктами окислительного присоединения, имеющими иодид как лиганд. А в этой реакции это очень важно, потому что если CO не успевает внедриться, мы получаем продукт обычного кросс-сочетания. Для бромпроизводных и трифлатов необходима добавка KI как раз для того, чтобы заменить лиганд на палладии и ускорить внедрение CO.
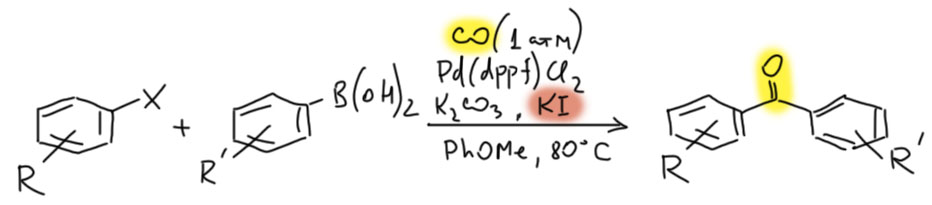
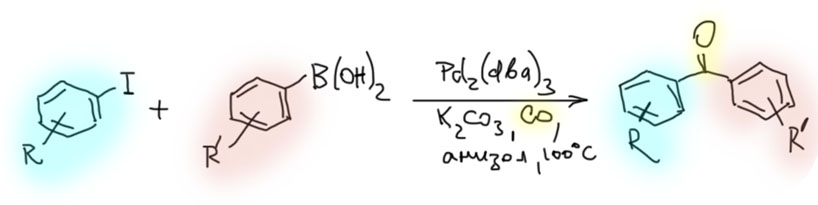
Реакция Мидзороки-Хека очень трудно сопрягается с карбонилированием - совсем не хватает места в координационной сфере палладия, да и олефин и CO конкурируют за место и активно мешают друг другу. Очевидно, что для успеха такой реакции требуется тщательный подбор контролирующего лиганда, но из-за толчеи и давки это очень непросто. Неудивительно, что в отличие от карбонилирующего кросс-сочетания, известного почти столько же сколько и само карбонилирование, карбонилирующий Хек представлял собой почти нерешимую задачу, пока за дело не взялся один из самых плодовитых исследователей в химии переходных металлов, Маттиас Беллер (ФРГ). Разработанный им метод использует трифлат к качестве субстрата, освобождающий одно место и позволяющий использовать бидентатный лиганд dppp (Beller M. et al Angew. Chem. Int. Ed. 2010, 49, 5284 –5288).
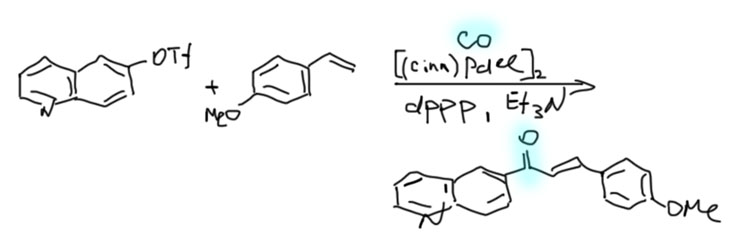
Структура продуктов однозначно свидетельствует о том, что внедрение CO происходит после окислительного присоединения, но до карбопалладирования - карбонилирование предшествует собственно реакции Мидзороки-Хека.
Более сложные каскады могут включать много стадий в едином каталитическом цикле...
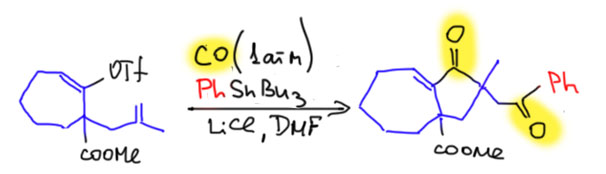
... как например, в каскаде, начинающемся как карбонилирование, но развивающемся дальше через циклизацию в результате внутримолекулярного карбопалладирования, за которым следует ещё одно карбонилирование, и завершающимся кросс-сочетанием по Стилле с оловоорганикой.
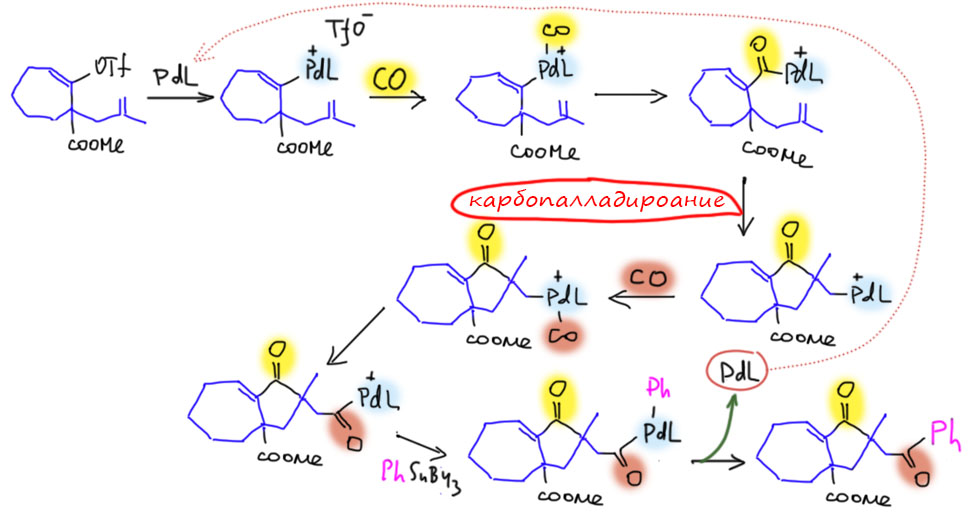
Кетоны и всякие производные карбоновых кислот получать карбонилированием мы уже научились. Но хотелось бы ещё альдегиды. В стехиометрической реакции с реагентов Коллмена ацильный комплекс расщепляют протолизом. Но в каталитических реакциях удобнее было бы расчитывать на восстановительное элиминирование, но для этого нужно загрузить в координационную сферу и гидрид и ацил. Один из способов это сделать очень сильно похож на карбонилирующее кросс-сочетание, но на последней стадии на металл загружается гидрид переметаллированием с гидридом кремния (силаном).
Результат - весьма хемоселективная реакция - обратите внимание, что сохраняется даже олефиновая двойная связь, для которой в этой реакции очень много угроз - и гидрирование, и Хек - но ни одна не мешает целевой реакции гидрокарбонилирования.
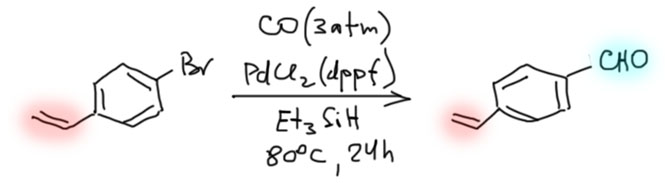
В каталитическом цикле после окислительного присоединения формируется ацильный комплекс, который в восстановительном элиминировании даёт формильную группу. В принципе, это тоже каскад, хотя и простой.
А теперь займёмся очень сложной химией, в которой карбонилирование сочетается с реакциями двойных и тройных связей. Здесь много важнейших промышленных реакций, ещё больше трудноразрешимых проблем, и как минимум, одна, но чрезвычайно популярная в тонком органическом синтезе реакция Посона-Кханда. И здесь уже мало палладия, и много металлов 9 группы кобальта и родия.
Знаменитая и чрезвычайно важная промышленная реакция гидроформилирования олефинов или реакция Рёлена - случайный побочный продукт лихорадочных попыток найти способ получения синтетического топлива в нацистской Германии. Для этого там интенсивно исследовалась реакция Фишера-Тропша - процесс синтеза предельных углеводородов из синтез-газа. Процесс работал и снабжал Третий Райх значительным количеством довольно низкокачественного эрзац-бензина, но в попытках усовершенствовать метод Отто Рёлен пробовал добавлять в синтез-газ простые олефины, и обнаружил образование альдегидов. Даже эту реакцию пробовали приспособить для получения взрывчатых веществ из аналогов пентаэритрита, но не преуспели. После войны исследование процесса возродилось уже именно для синтеза промышленно важных альдегидов.

Otto Roelen
(1897-1993)
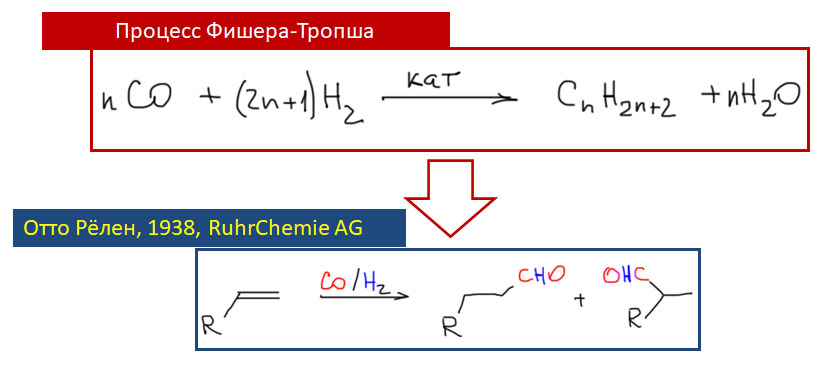
Каталитический цикл гидроформилирования был в основном установлен в работах Ричарда Хека. Можно даже сказать, что именно с этого и начался весь катализ комплексами переходных металлов в органической химии. Каталитический цикл можно представить себе как гидрирование в середину которого вставили миграционное внедрение карбонила.
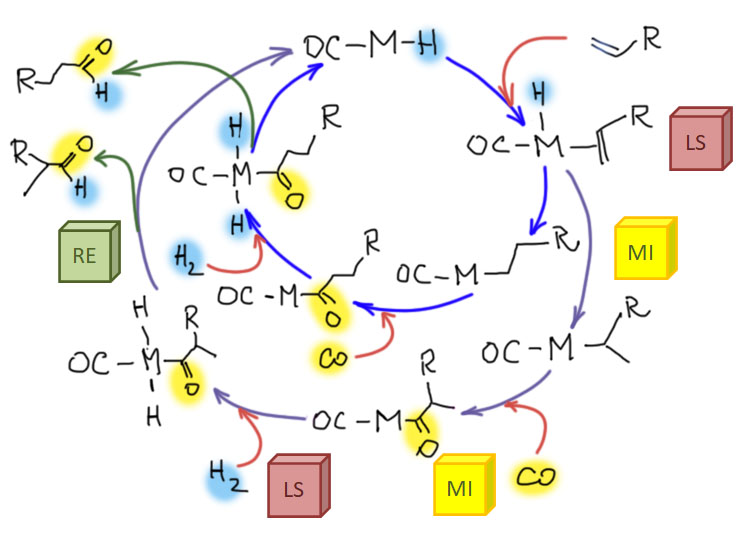
Проблема гидроформилирования в том, что в несимметричных олефинах, а реакцию чаще всего делают с терминальными олефинами, получается два продукта, называемые по аналогии марковниковким и анти-марковниковским (или даже чаще говорят про альдегиды нормального и изо-строения). Направление реакции определяется первой стадией - миграционным внедрением гидрида в олефин. Фокус в том, что в разных областях нужны альдегиды и того и другого строения, и поэтому исследования региоселективности гидроформилирвоания стали важнейшей научной задачей, решение которой принесло огромное количество знаний о функционировании каталитических систем.
Первые катализаторы гидроформилирования используют карбонильные комплексы кобальта. Простой тетракарбонилгидрид, образующийся на месте из карбонила кобальта даёт смесь лдегидов нормального и изо-строения с преобладанием первого, но недостаточно большим. Региоселективность сильно улучшается, а условия реакции смягчаются, если в реакцию вводят фосфиновые лиганды.
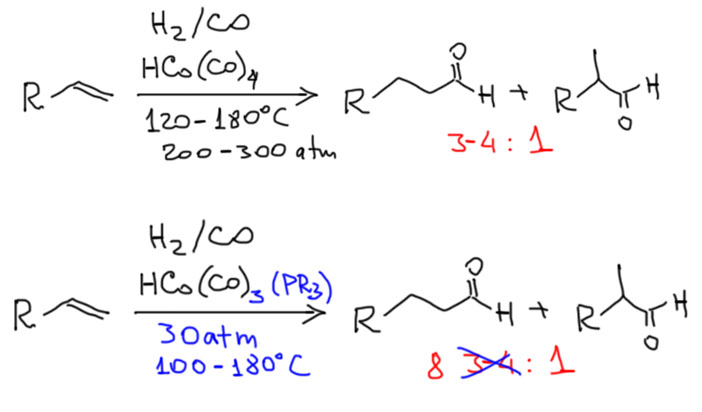
Региоселективность реакции определяется в обычном для присоединения гидридов металлов процессе обратимого присоединения - гидридного отщепления, что вызывает блуждание двойной связи, на фоне которого и происходит миграционное внедрение карбонила. Более стерически объёмистый кобальт с фосфиновым лигандом и мигрирует быстрее, и в большей степени предпочитает терминальное положение.
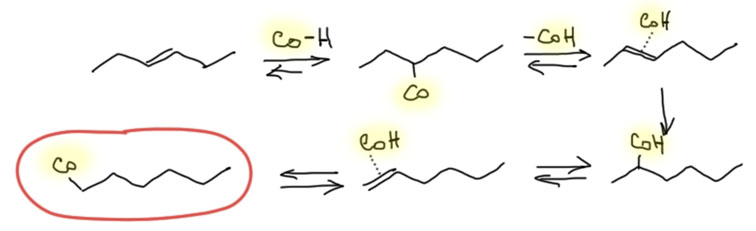
При использовании родиевых катализаторов тенденции в региоселективности те же, но ещё более выражены - фосфиновые комплексы дают ещё больший выход альдегидов нормального строения. Именно поэтому родиевые комплексы чаще применяют в промышленных процессах.
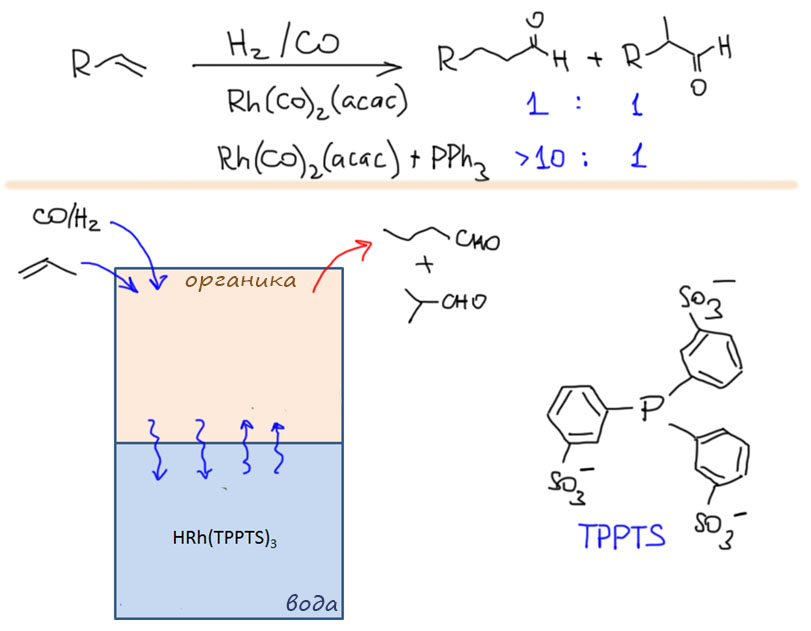
Н-масляный альдегид очень востребован в промышленности, как важный синтетический полупродукт. Для получения этого соединения в больших масштабах компании РурХеми и Рон-Пуленк совместно разработали эффективный процесс, использующий фазо-разделяемый катализ с рециклизацией катализатора в водной фазе. Водорастворимость катализатора достигается использованием уже нам знакомого сульфированного трифенилфосфина (собственно именно для этого процесса он и был впервые разработан).
Как показали исследования, реакция идёт в водной фазе из-за весьма приличной растворимости маленького олефина пропилена в воде. Продукты экстрагируются в органическую фазу и отделяются от каталитической фазы. Более крупные и более гидрофобные олефины в этой системе не реагируют, так как совсем нерастворимы в воде.
Гидроформилирование, как и другие фундаментальные превращения, можно встраивать в более сложные процессы. Познакомимся с ещё одним принципом - не с каскадированием, а с тандемным процессом. Тандем - это последовательность разных реакций, имеющих разные механизмы и не встроенных в один каталитический цикл с единым катализатором.
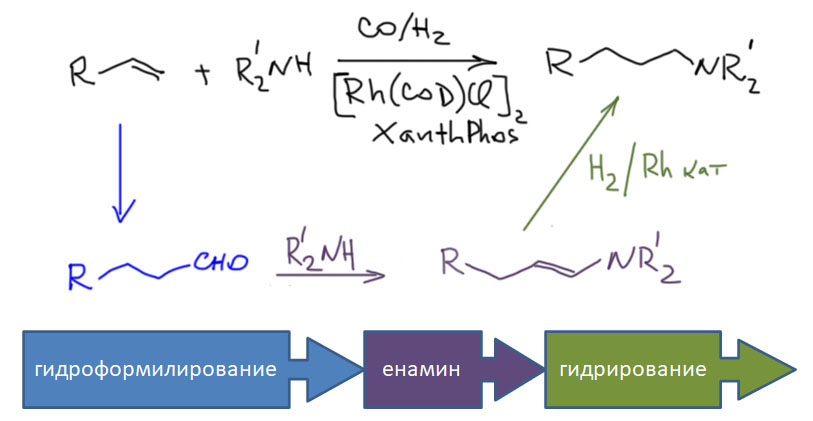
Соединяем гидроформилирование, дающее альдегид, с обычным образованием енамина с вторичным амином, за которым следует гидрирование и опять с родиевым катализатором - можно сказать тем же, что и в гидроформилировании, но дело в том, что он ведёт отдельный цикл гидрирования, отделённый от гидроформилирования стадией образования енамина, совсем не нуждающейся в родии (родию должно быть обидно, но ничего - потерпит).
Одна из самых популярных и важных реакций с участием комплексов переходных металлов в органическом синтезе - реакция Посона-Кханда. Питер Посон - это тот самый учёный, который впервые получил, вместе с Томасом Кили, ферроцен. Этого достаточно, чтобы навсегда войти в историю химии, но ему повезло ещё раз - повезло с пакистанским постдоком Ихсаном Уллой Кхандом, открывшим совершенно новую и очень интересную реакцию. Посон оказался исключительно щепетильным человеком, и не только не присвоил себе честь открытия, но и всячески после напоминал, что реакцию открыл отнюдь не он.

Peter Pauson
(1925-2013)
Реакция представляет собой циклизацию [2+2+1] олефина, ацетилена и карбонила из карбонила кобальта. Продукты реакции - циклопентеноны, превосходные заготовки для дальнейшего развития сложного синтеза. Реакция завоевала огромную популярность в органическом синтезе.
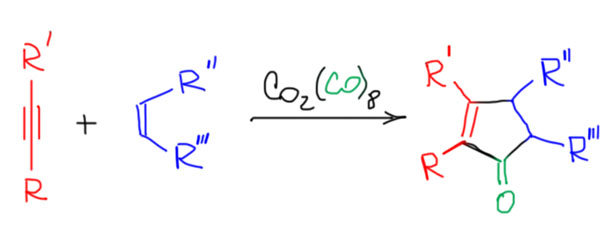
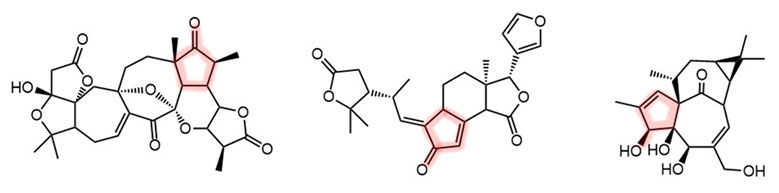
Циклопентаноновые и циклопентеноновые фрагменты очень часто встречаются в сложных природных и биологически активных молекулах, и реакция Посона-Кханда очень часто является ключом к синтезу таких соединений
Механизм реакции Посона-Кханда не менее красив, чем сама реакция Посона-Кханда. Во-первых, мы впервые встречаемся с реакцией, в которой работает не просто комплекс переходного металла, а двухядерный комплекс, причём работает согласованно обоими атомами металла.
Ключевой интермедиат реакции - комплекс ацетилена с карбонилом кобальта удивительной структуры - каждый атом кобальта образует η2-комплекс с одной из пи-связей ацетилена. Получается такой искажённый тетраэдр - ацетилен и два атома кобальта образуют два взаимно перпендикулярных треугольника. Так как в этом комплексе есть всязь металл-металл - это настоящий кластер.
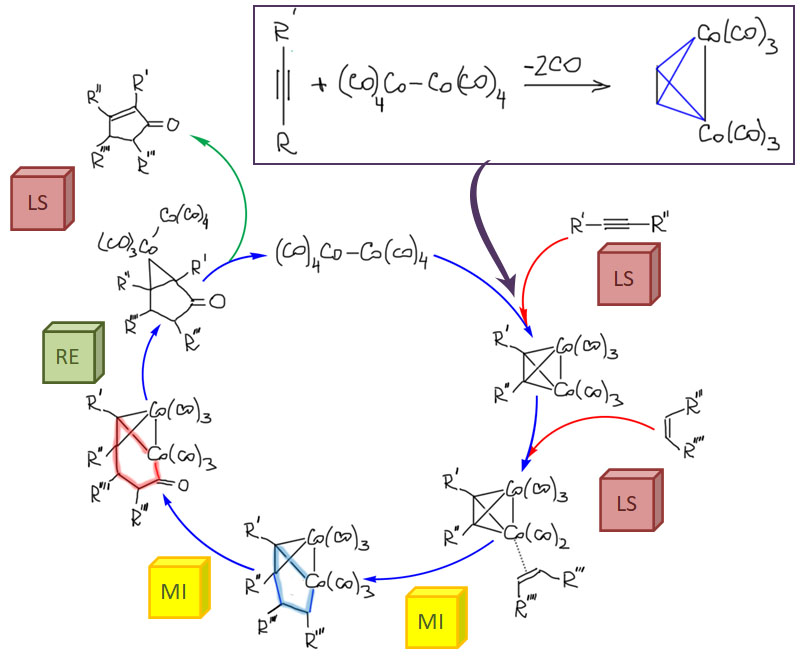
Механизм реакции Посона-Кханда похож на идеально отлаженную машину, в которой двухкобальтовый манипулятор поочерёдно берёт молекулы ацетилена, олефина и оксида углерода и филигранно соединяет их в новую молекулу.
И вот этот кластер, в котором один атом кобальта продолжает держать бывший ацетилен за одну пи-связь, другим атомом кобальта берёт олефин и миграционно внедряет его, образуя металлацикл. Дальше тот же атом кобальта ещё миграционно внедряет один из своих карбонилов. В каталитическом варианте реакции оксид углерода в конце снимает карбонил кобальта с двойной связи образовавшегося циклопентенона.
Диоксид углерода в смысле доступности и опасности отличается от CO радикально. Колоссальные количества этого вещества находятся в атмосфере и ежесекундно вырабатываются при сжигании топлива и жизнедеятельности живых организмов. Проблема глобального потепления как раз и связана с значительным повышением концентрации диоксида углерода в атмосфере по причине деятельности человека. Любители спорить с этим как-то совершенно не учитывают тот очевидный факт, что человечество за сто с небольшим лет достало из недр земли и вернуло в атмосферу то, что в этих недрах накапливалось миллионами лет, уходя как раз из атмосферы за счёт фотосинтеза и захоронения растительных остатков на больших глубинах. Тут как раз неплохо бы не доказывать, почему в атмосфере так сильно возрос уровень углекислоты, а желающим это опровергнуть – попробовать объяснить, куда подевался такой чудовищный объём возвращённого в атмосферу углекислого газа, и как может такое быть, что такое кратковременное изменение состава атмосферы вообще не имеет последствий. А объём действительно чудовищный, ведь львиная доля потребностей всего человечества в энергии была покрыта за счёт сжигания ископаемого топлива.
Многим приходит в голову идея, что углекислый газ нужно из атмосферы забирать. А куда девать? Самое надёжное было бы отправить обратно в глубины земли. Но как это сделать совершенно непонятно. И поскольку человек – существо недальновидное, но промысловатое, рождаются идеи как бы не просто углекислый газ из атмосферы забирать, но ещё что-то полезное из этого делать, что можно хорошо продать и тем самым покрыть расходы на это производство, да еще и наварить немного. Получить пока из CO2 ничего особенного не удалось, но саму эту идею удалось хорошо продать политикам разных стран и чиновникам международных организаций так, что исследования в этой области в последние 10 лет стали финансировать не просто щедро, а прямо баснословно щедро. В научных журналах последних 10 лет просто глаза рябит от статей про новые реакции из CO2, и в начале почти каждой такой статьи будет обязательно написано про благородную цель избавить человечество от медленного поджаривания и одновременное получение ценнейших материалов, лекарств, полимеров, и т.п. Большинство таких исследований производят совершенно комическое впечатление. Что-то полезное в них подчас действительно получается, но авторы умудряются не замечать двух вещей: мизерных потребностей человечества в предлагаемых продуктах и огромных затрат материалов и энергии на осуществление предлагаемой реакции. Проблема в том, что CO2 из атмосферы нужно убирать в масштабах десятков гигатонн, а всё производство органической продукции химической промышленности оценивается величинами в сто раз меньшими. Поэтому даже в совершенно фантастическом варианте, когда вообще все органические материалы будут получать из CO2, количество забранного из атмосферы на эти цели диоксида углерода было бы пренебрежимо мало и на баланс углерода серьёзного влияния бы не оказало. Точность современных оценок этого баланса гораздо больше, чем эти количества, и титанических усилий планета наша просто не заметила бы. Но, увы, это ещё не всё. На любой химический процесс тратится очень много энергии и материалов. В расчётах все такие затраты пересчитывают на затраченную энергию, и делают простую оценку, сколько CO2 при этом будет выброшено обратно в атмосферу – это называется “углеродным следом” продукции или процесса. Проблема ведь в том, что энергию до сих пор в основном получают сжиганием ископаемого топлива. Можно просто взять энергетические и топливные балансы крупных стран, чтобы в этом убедиться. Да, когда-нибудь энергию будут получать целиком из возобновляемых источников с нулевым углеродным следом. Но тогда и выброс CO2 в атмосферу уменьшится действительно радикально, планета придёт к новому балансу, и смысла забирать из атмосферы углекислоту не будет.
Очень забавный пример уровня таких рассуждений – часто приводимый пример того, что диоксид углерода уже используется в больших масштабах в промышленности – производство карбамида, то есть мочевины из аммиака и углекислого газа. Это действительно очень крупномасштабный процесс. И мочевина совершенно официально считается органическим соединением, и даже более того – первым органическим соединением, полученным синтезом из более простых, официально неорганических соединений. Считается, что именно с этой реакции, реакции Вёлера, идёт вся органическая химия как наука. Это всё очень трогательно, если забыть, для чего используется получаемая таким образом мочевина. Но – это удобрение, азотное удобрение, очень хорошее. И первое, что с ним происходит, когда оно попадает в почву – расщепление микроорганизмами на ион аммония и – диоксид углерода. Это Бог дал – Бог и взял. А у нас всё наоборот – углекислый газ взяли – углекислый газ и отдали. Ну, хоть растения растут, но CO2 вернулся в атмосферу. На самом деле даже в гораздо больших количествах, чем взяли, потому что аммиак, необходимый для этой реакции, образуется очень энергозатратным синтезом Габера-Боша, имеющим очень большой углеродный след.
Всё это безусловно не значит, что заниматься химией CO2 не стоит. Химией вообще заниматься стоит, но именно с целью получения полезных материалов и веществ. Если будет хороший процесс с использованием углекислоты – очень хорошо. Пока с этим не очень хорошо, но нас должна вдохновлять Природа, которая очень хорошо справляется с CO2 – для неё это основной источник углерода (carbon feedstock) для синтеза всех органических соединений, и это не только фотосинтез, но и довольно многочисленные реакции ферментативного карбоксилирования.
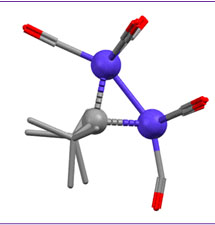
Диоксид углерода - очень непростая молекула, у неё не так-то много возможностей для реакций и для активации. Одна из её проблем - очень высокая термодинамическая устойчивость (стандартные энтальпия и свободная энергия образования отрицательны и очень велики ΔHº=-94 ккал/моль, ΔGº=-109 ккал/моль при 298 К) и это даёт плохой вклад в любые равновесия с участием диоксида углерода - они чаще бывают смещены к исходным, чем к продуктам. Иными словами, в химии гораздо больше реакций декарбоксилирования (потери CO2, которая является очень хорошей уходящей группой электрофугного типа), чем карбоксилирования.
Диоксид углерода - электрофил по атому углерода. На первый взгляд, это должен быть очень хороший электрофил - углерод между двумя электроотрицательными атомами, или два взаимно перпендикулярных карбонила - любой такой способ посмотреть на структуру вроде бы говорит об том. Но не стоит забывать две вещи - во-первых, углерод имеет sp-гибридизацию, а следовательно и относительно более высокую эффективную электроотрицательность. Во-вторых, в каждой из перпендикулярных пи-систем карбонил сопряжён с неподелённой парой второго кислорода, образуя нечто типа карбоксила, а не карбонила.
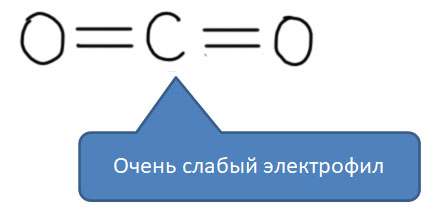
Самый основной способ активации молекулы CO2 - увеличение электрофильности за счёт координации с кислотой Льюиса. Такая координация разрушает сопряжение между кислородом и карбонилом, и делает углерод весьма компетентным электрофильным центром. С точки зрения нашего курса это не очень хорошие новости, потому что электрофильную активацию могут с тем же успехом оказывать и производные непереходных металлов, и действительно, среди активаторов диоксида углерода очень много солей и комплексов лития, магния, цинка, алюминия и их родственников в соответствующих группах за пределами d-блока.
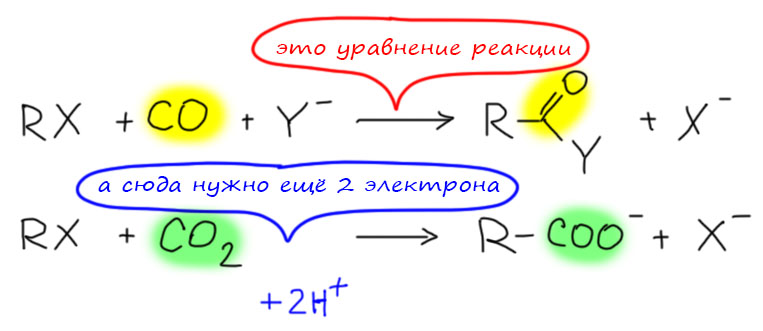
Карбоновые кислоты и их производные с точки зрения структуры, гораздо ближе к диоксиду углерода, чем к оксиду, но с точки зрения катализа это не так очевидно. Хорошо нам уже известные реакции нуклеокарбонилирования просто идеально приспособлены под катализ переходными металлами - стехиометрическое уравнение реакции пишется без дополнительных реагентов и уравнено: осталось найти катализатор.
А вот реакция с диоксидом углерода, во-первых, ведёт только к самой карбоновой кислоте, но не к её производным, а, во-вторых, не уравнивается без дополнительного источника электронов: катализа мало, нужен ещё восстановитель, а значит, мы удаляемся от принципа экономии атомов, а не приближаемся к нему.
Вот типичный пример карбоксилирования, описанный большим любителем этой химии каталонским исследователем Рубеном Мартином (T. León, A. Correa, R. Martin J. Am. Chem. Soc. 2013, 135, 4, 1221). Оцените высокую загрузку пред-катализатора (а значит, очень маленький TON), необходимость добавления не самого тривиального фосфинового лиганда (трициклогексилфосфин это вам не трифенилфосфин, на дороге не валяется, и требует к себе весьма трепетного отношения, как все триалкилфосфины, обычно самовоспламеняющиеся на воздухе).
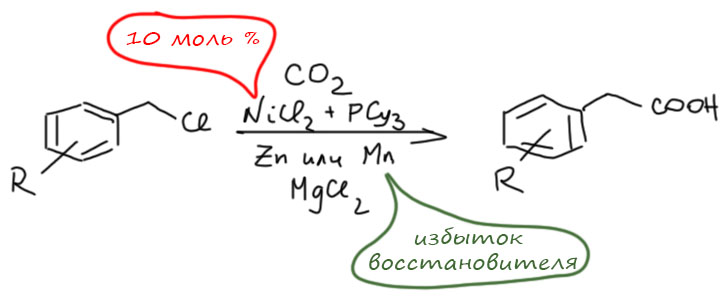
И необходимость добавления очень щедрого количества восстановителя, опять таки, обычно в кратном избытке просто потому что скорость реакции с поверхности сильно отстаёт от других стадий каталитического цикла, и с этим ничего не поделать, кроме как просто добавить побольше. Зато реакция идёт в очень мягких условиях, при комнатной температуре при обычном давлении CO2.
Результат налицо - если нам нужно превратить галогенпроизводное в карбоновую кислоту, мы почти наверняка предпочтём реакцию оксида, а не диоксида углерода. И это вполне подтверждается литературным поиском - если нуклеокарбонилирование очень популярно в реальных синтезах, то применения карбоксилирования мы пока не найдём ни одного.