Реакции кратных связей. Часть 2.
Реакции олефинов и ацетиленов с участием комплексов переходных металлов далеко не исчерпываются активацией кратных связей в разнообразных реакциях присоединения. Как и в обычной органической химии возможно участие в реакциях соседнего с кратной связью атома углерода – аллильной или пропаргильной системы. В химии переходных металлов аллильные реакции встречаются гораздо чаще, чем пропаргильные. Аллильные комплексы легко образуются и имеют разнообразную реакционную способность, как в каталитических, так и в стехиометрических реакциях.
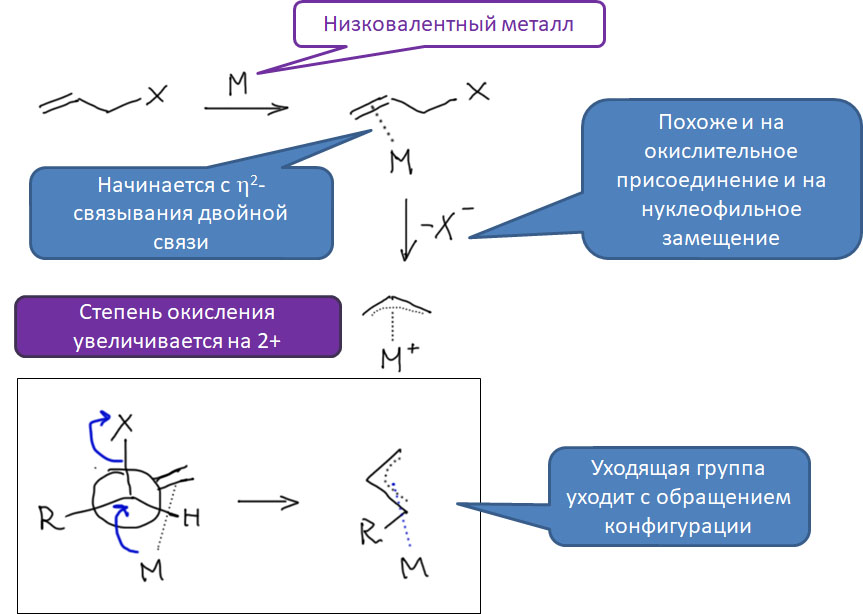
Поздние переходные металлы легко образуют η3-аллильные комплексы. Реакций, приводящих к таким комплексам очень много, но самая важная - окислительное присоединение к металлу аллильного электрофила: такую реакцию можно представить, как двустадийную, состоящую из образования η2-комплекса с двойной связью, за которым следует окислительное присоединение. Возможен и обратный порядок - окислительное присоединение, за которым следует перегруппировка σ-комплекса в η3-комплекс.
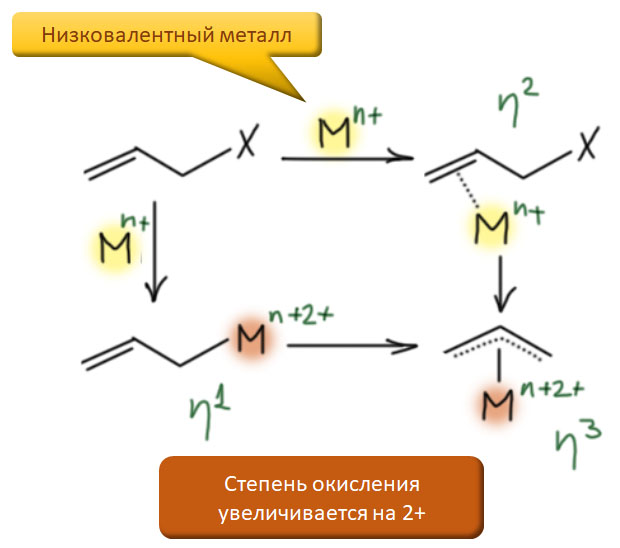
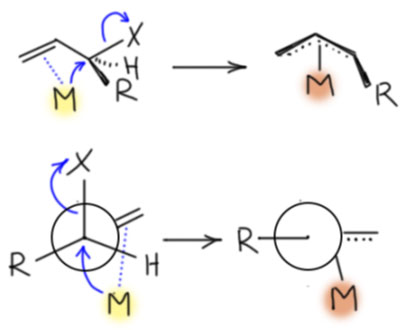
Образование аллильных комплексов в результате окислительного присоединения происходит стереоспецифически - металл подходит к аллильной системе со стороны, противоположной уходящей группе. Происходит обращение конфигурации, только его сложнее опознать, чем в обычном нуклеофильном замещении потому что аллильная система в образовавшемся комплексе плоская, но нужно учесть и металл, который "повисает" на аллиле с обратной стороны и уничтожает плоскость симметрии - это хорошо видно на проекции Ньюмена. К сожалению, из-за спонтанной изомеризации η1-η3, многие аллильные комплексы конфигурационно неустойчивы, но с этим можно справиться, и это открывает возможности применения этих комплексов в стереоселективных реакциях.
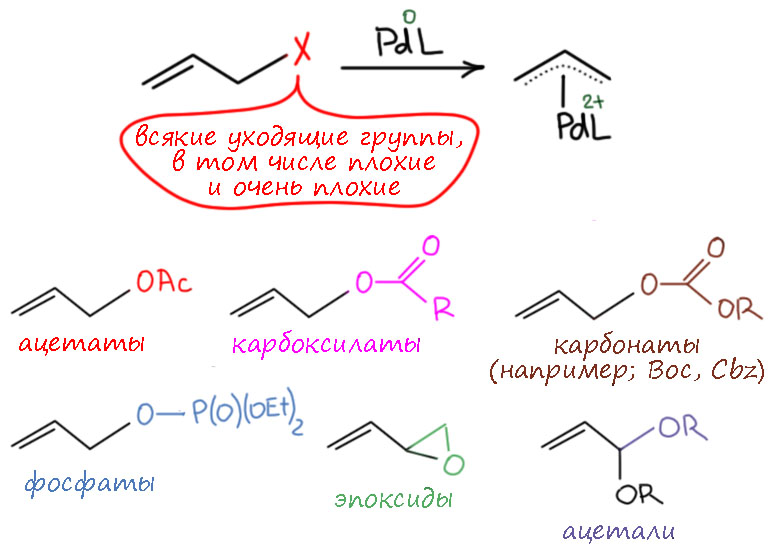
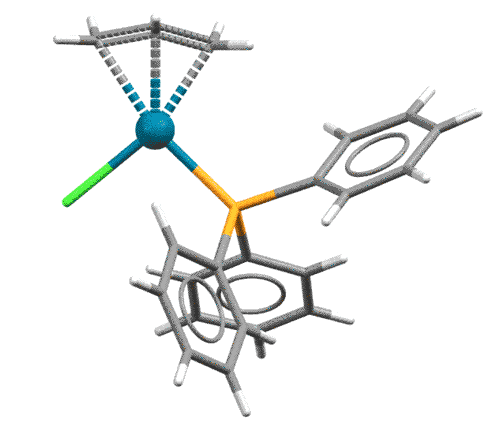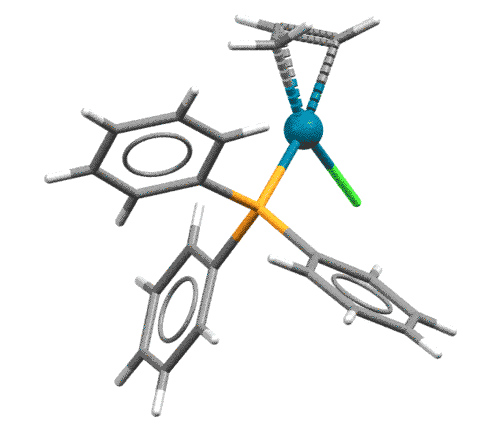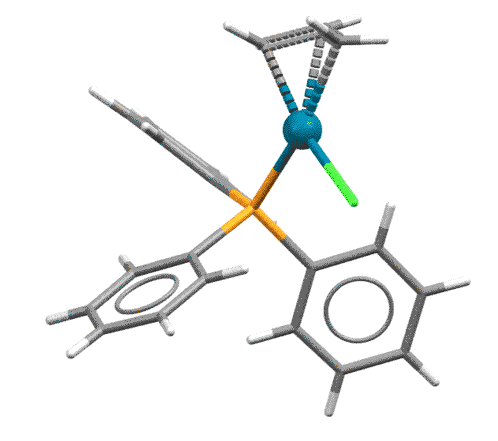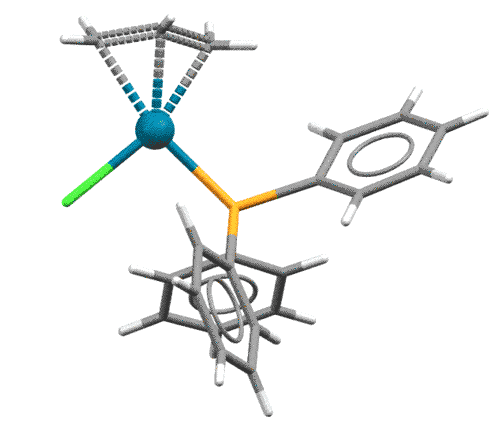
Особенно интересны и очень хорошо изучены комплексы палладия. Такие комплексы легко образуются из разнообразных аллильных производных, причем даже с такими уходящими группами (ацетат, другие карбоксилаты, карбонаты, даже просто спирты), которые никогда не участвуют в обычном окислительном присоединении к Pd(0). Очевидной причиной такой реакционной способности является дополнительный выигрыш от образования связей в тригапто-комплексе.
Аллильный лиганд несколько отличается по структуре от полностью плоских аллильных аниона и катиона - заместители в нем отклоняются от плоскости, в которой лежит треугольник углеродов аллила, в сторону, противоположную от атома палладия. Отклонения невелики, и с точки зрения стереохимии аллил можно считать плоским с металлом, который присоединён с одной стороны от плоскости, и это имеет очень важные последствия для стереохимического анализа реакций аллильных комплексов.
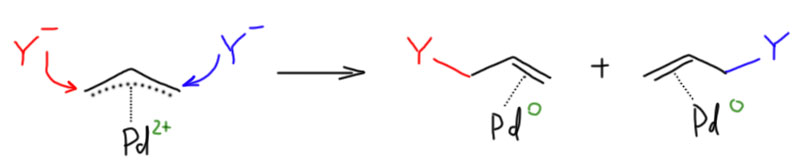
Основная реакция аллильных комплексов - нуклеофильная атака на η3-аллильный лиганд. Это довольно странное свойство для металлоорганического соединения открыл Дзиро Цудзи с сотр. в 1965. Металлоорганика обычно нуклеофильна и реагирует с нуклеофилами, а здесь наоборот. Но вряд ли это так уж удивительно, ведь мы уже знаем, что переходный металл не в самой низкой степени окисления обычно оперирует как электрофильный активатор, кислота Льюиса. А у палладия их, в принципе, вообще и есть две: ноль как низковалентная форма и 2+ как высоковалентная (есть ещё 4+, но это большая редкость, и 1+, но это пока экзотика), поэтому Pd(2+) работает как электрофильный активатор, стягивая с лиганда электронную плотность и активируя его к реакции с нуклеофилами.
При атаке нуклеофила на η3-аллильный лиганд палладий забирает два электрона, и переходит в Pd(0) в виде слабого η2-комплекса. Важно то, что нуклеофил может атаковать любой из крайних атомов аллильной системы, так что в таких реакциях с участием несимметричных или изотопно-меченых аллилов получается два продукта. В конкретных реакциях преимущественное направление замещения определяется в основном стерическими факторами, хотя иногда работают и электронные.
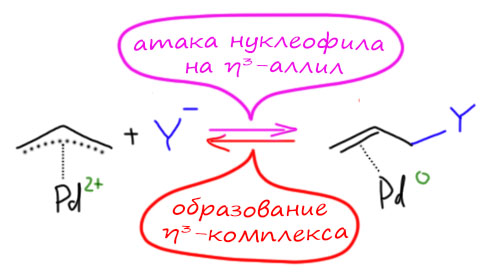
Как и многие другие реакции с участием переходных металлов, и эта реакция представляет собой формально обратную пару к другой реакции - в данном случае это и есть реакция образования η3-аллильного комплекса. Нуклеофил в одной реакции становится уходящей группой в другой. И мы ещё с обычной органической химии знаем правило - хорошая уходящая группа - это плохой нуклеофил, и наоборот. Поэтому для образования комплекса мы будем выбирать хорошие уходящие группы (правда немного не в том смысле, как тот, к которому мы привыкли, например, в SN2. И для атаки нуклеофила мы будем выбирать то, что уходящей группой уже не станет.
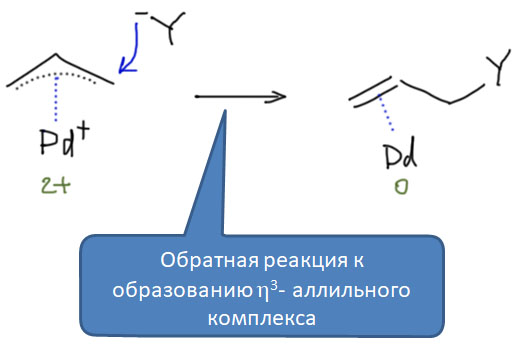
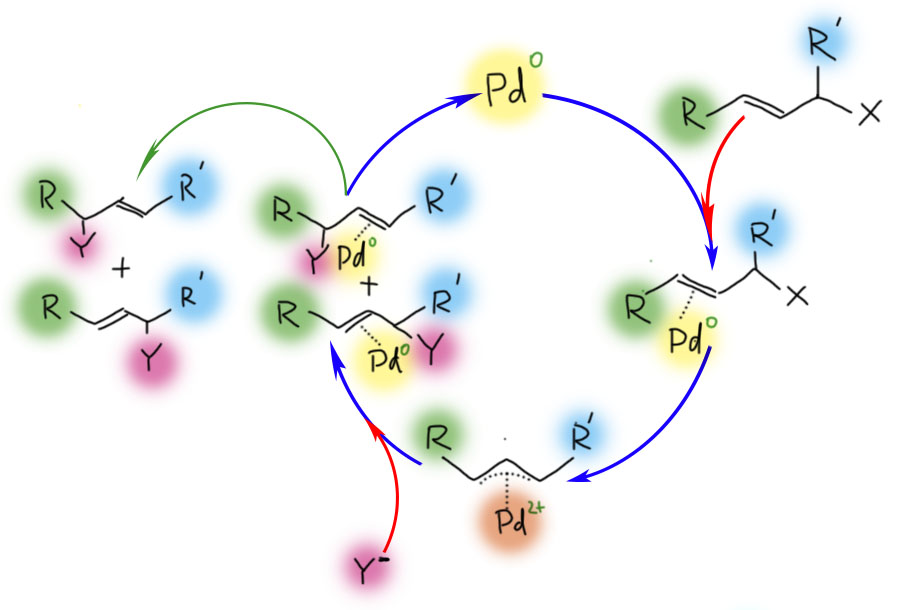
Основная реакция аллильных комплексов - нуклеофильное замещение. Это довольно странное свойство для металлоорганического соединения открыл Дзиро Цудзи с сотр. в 1965. Металлоорганика обычно нуклеофильна и реагирует с электрофилами, а здесь наоборот. Более того, и аллильные комплексы, если они не тригапто, а моногапто-связаны, также нуклеофильны. Именно тригапто-связывание приводит к обращению реакционной способности, к электрофильной активации аллила, который ведет себя в комплексе , как аллильный катион.
Последовательность реакций образования аллильного комплекса и нуклеофильного замещения может быть выполнена как каталитическая реакция. Так же как в обычном аллильном замещении каталитическое аллильное замещение может давать два продукта - прямого замещения и замещения со сдвигом двойной связи (аллильной перегруппировкой). В современной химии реакцию называют именами Цудзи, который её открыл, и Троста, который разработал ее энантиоселектвный вариант, завоевавший большую популярность в синтезе.
- Гораздо шире выбор нуклеофилов
- Не нужно готовить нуклеофил заранее – годится и сопряженная кислота
- Разная селективность катализируемой и некатализируемой реакций
- И самый главный бонус - энантиоселективность
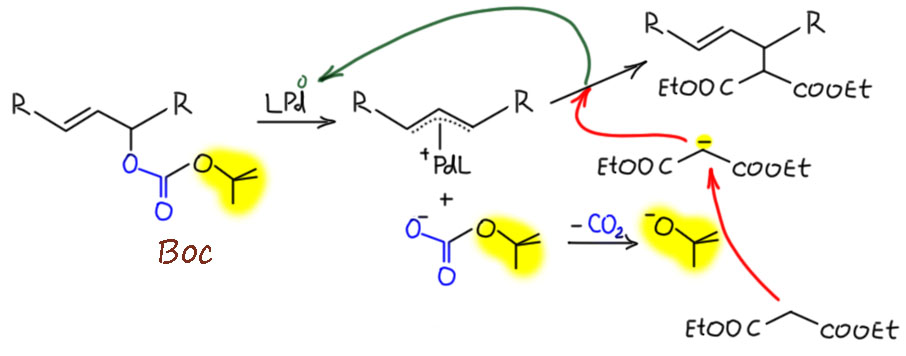
Обычное нуклеофильное замещение в аллильных системах это, в зависимости от условий, или SN2 или SN1-замещение, со всеми вытекающими проблемами и ограничениями. Катализируемое аллильное замещение отлично работает для гораздо более широкого круга нуклеофилов, имеет другие закономерности, в том числе и с точки зрения аллильных перегруппировок, и т.д. Самая главная особенность каталитического замещения, объясняющая большую популярность этого метода в синтезе, состоит в возможности энантиоселективной реакции с третичными аллильными субстратами с переносом хиральности от катализатора, от хирального лиганда.
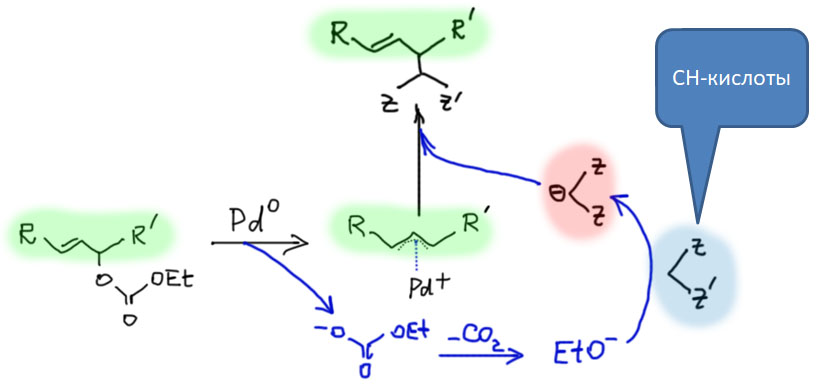
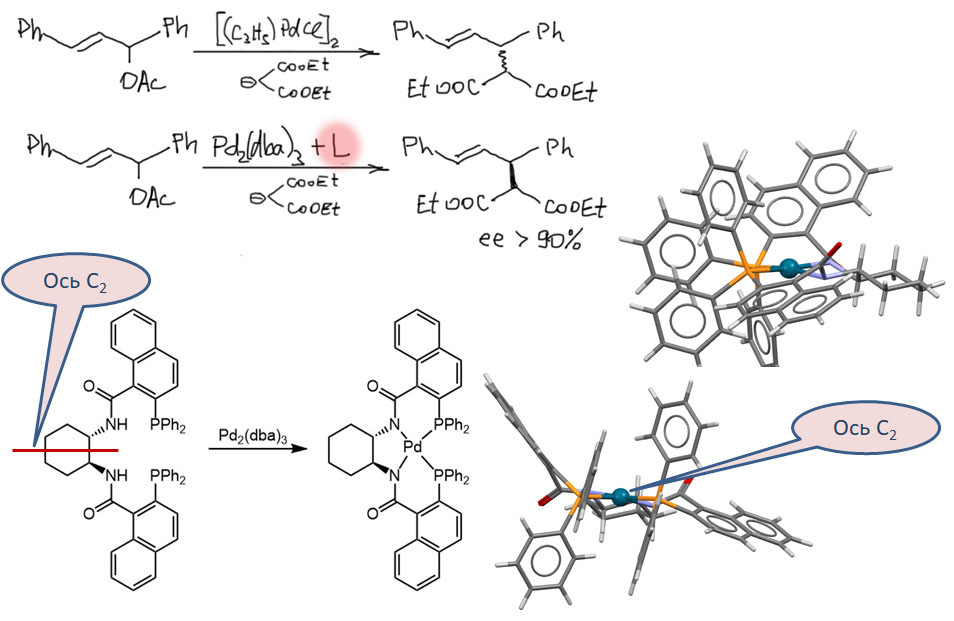
Энантиоселективный вариант аллильного замещения часто называют реакцией Цудзи-Троста по именам исследователей, которые обнаружили аллильное замещение (Цудзи) и разработали его асимметрическую версию (Трост). Трост же разработал первый эффективный лиганд - тетрадентатный хелатор, смонтированный на хиральной молекуле транс-1,2-диаминоциклогексана, который так и называют лигандом Троста. Первоначальное объяснение работы этого комплекса предполагало образование жёсткого тройного хелата с сильно асимметричной структурой. Выглядит красиво, но категорически непонятно, как в таком комплексе может происходить аллильное замещение - где в нём возможности для связывания аллила. Дальнейшие исследования показали, что всё гораздо сложнее, и что красивая концепция обязательности C2-симметричного хирального окружения атома металла далеко не всегда работает.
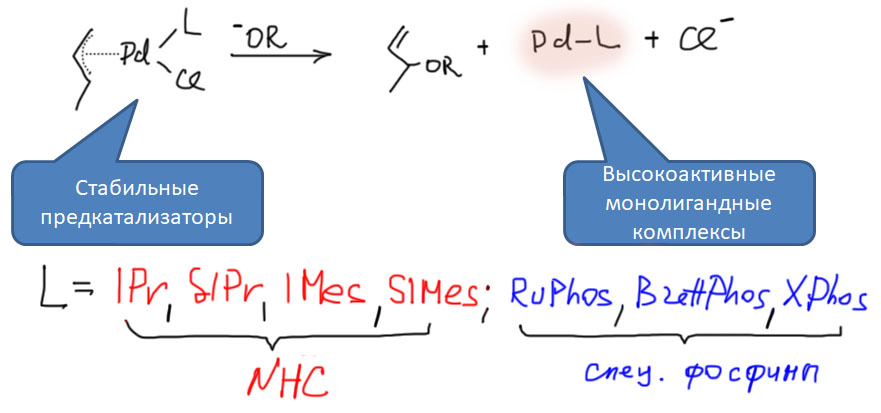
Образование нульвалентного палладия при реакции аллильных комплексов палладия с нуклеофилами нашло применение для генерации высокоактивных монолигандных комплексов - катализаторов кросс-сочетания. Этот метод особенно привлекателен для реакций Судзуки-Мияуры и Бачуолда-Хартвига, в которых используются основания, которые прекрасно справляются и с ролью нуклеофилов для пред-активации. Таким способом генерируют комплексы палладия с гетероциклическими карбенами (NHC) и фосфинами, особенно специальными лигандами на основе дифенила и ферроцена.
Использование таких пред-катализаторов дает высокоактивные каталитические системы для проведения реакций с малореакционноспособными субстратами, в мягких условиях.
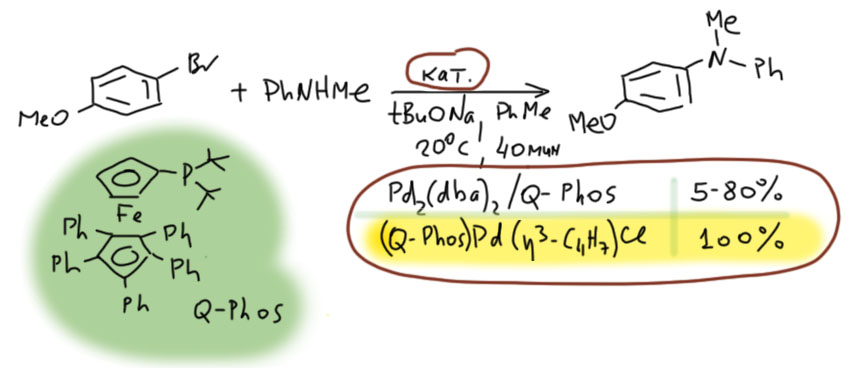
Например, в арилировании вторичного амина малоактивным бромпроизводным использование специального лиганда Q-Phos третьего поколения даёт высокоактивную систему и реакция идёт при комнатной температуре и очень быстро, - но с плохой воспроизводимостью при препаративных количествах реагентов. Использование пред-катализатора аллильного типа с тем же лигандом даёт ещё больший выход с высокой воспроизводимостью.

Если двойную связь олефина разорвать целиком, то получатся два карбена. Такая реакция, конечно, в обычных условиях не происходит, но формально это именно так. Карбены находятся в таком же отношении к алкенам, как свободные радикалы – к алканам. В обычной органической химии карбены встречаются довольно часто, но почти всегда как высокореакционноспособные интермедиаты, участвующие в различных реакциях. Время жизни большинства свободных карбенов мало, они не могут быть выделены, исследованы не в виде молекулы в специфических условиях (высоком вакууме, криогенных матрицах и т.п.), а в виде вещества, которое можно было бы охарактеризовать обычными физико-химическими и спектральными методами. Тем не менее, карбен можно стабилизировать правильно подобранными заместителями, и такие стабилизированные или даже просто стабильные карбены появились в органической химии в 1990-х, и с тех пор приобрели невероятную популярность во всех частях органической химии.
В химии переходных металлов есть очень глубоко укоренившаяся нехорошая привычка называть карбенами комплексы с участием карбеновых лигандов. Например, говорят про карбены Фишера, карбены Шрока, родиевые карбены – имея в виду не собственно карбены, а некие специфические типы комплексов. Нужно очень аккуратно к этому относится, и всегда, читая статьи или слушая доклады, добиваться понимания того, что имеется в виду – комплекс или сам лиганд. Главный признак карбеновых лигандов очень прост, в одном из лигандов связанный с металлом углерод должен иметь два и только два связанных с собой атома (быть двухвалентным, если мысленно оторвать эту группу от металла). Это очень формальный признак, ничего не говорящий о саой химической природу и реакционной способности такого лиганда – он может быть чрезвычайно сильным электрофилом или очень сильным нуклеофилом, или вообще не проявлять никакиз признаков собственной реакционной способности. В этом есть очень большая сложность, потому что никогда нельзя сказать из общих соображений, чего мы можем ожидать от того или иного комплекса с карбеновым лигандом – нужно обязательно разобраться с деталями строения и взаимного влияния металла, карбена и анциллярных лигандов.
Но в химии переходных металлов карбеновые комплексы, которые бывают и совершенно стабильными и настолько нестабильными, что поймать их не легче, чем сами простые карбены, получили широчайшее распространение. Есть несколько больших типов карбеновых комплексов, и карбеновые лиганды могут служить как лиганды-акторы, так и очень интересные анциллярные лиганды.
Этот слайдер обновлён 12.04.2022. Новые или обновлённые слайды можно легко узнать по анимированным заголовкам и эффекту инверсии при наведении курсора на заголовок.
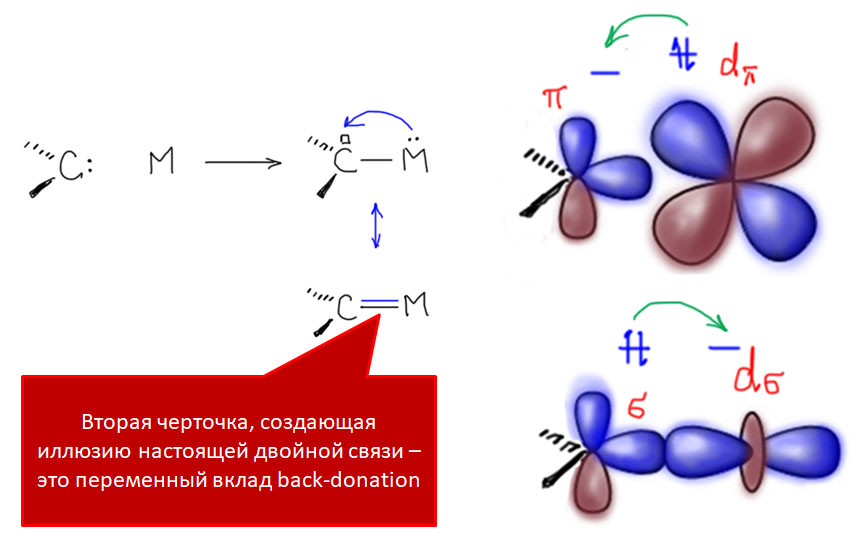
Карбены, как правило, неустойчивые и высокореакционноспособные частицы в свободном состоянии, но они же могут служить лигандами для переходных металлов. Большинство карбенов - типичные нейтральные L-лиганды, не изменяющие степень окисления металла. Связь металла с карбеном обслуживается координационной связью за счет неподеленной пары углерода карбена. Дополнительно металл может воспользоваться пустой орбиталью карбена для back-donation. Эту дополнительную связь часто обозначают второй черточкой, как будто карбен с металлом связан двойной связью. Но вклад back-donation очень сильно различается в различных карбеновых комплексах, и это обстоятельство необходимо иметь в виду при анализе структур конкретных комплексов.
После открытия первых карбеновых комплексов в 1964 году, этот тип лигандов сначала понемногу, но чем дальше тем настырнее, вторгался в координационную и органическую химию. Выяснилось, что существует очень много разных типов таких лигандов, что их стабильность и реакционная способность очень сильно зависит и от структуры органической части, и от металла, и от других лигандов на металле. Современная химия карбеновых комплексов ошеломляюще сложна, они участвуют в огромном количестве каталитических и стехиометрических реакций, в том числе таких знаменитых и популярнейших реакций как метатезис олефинов и CuAAC-клик реакция, но и это только малая доля.
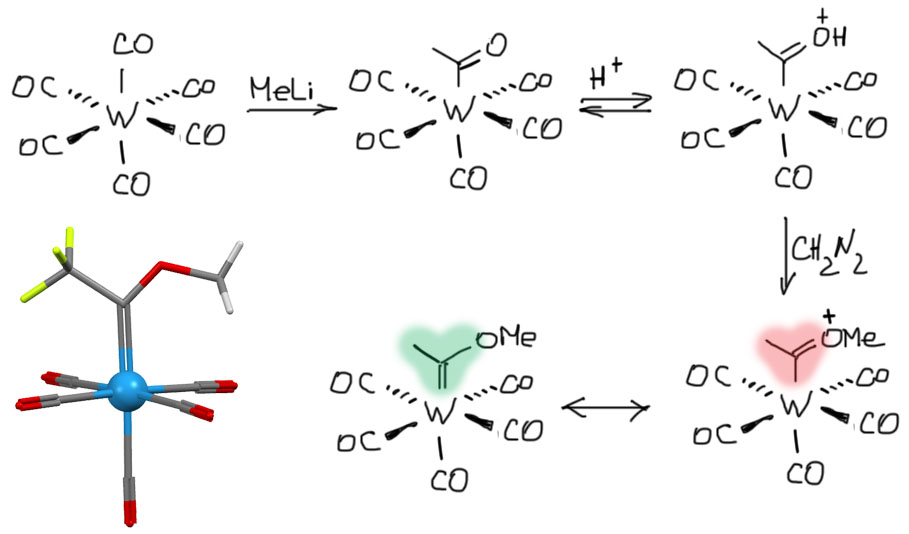
Первый комплекс лиганда, формально соответствующего понятию карбен, то есть имеющему углерод, связанный только с двумя группами помимо металла, получил в 1964 г. Эрнст Отто Фишер (нобелевский лауреат вместе с Уилкинсоном за определение структуры ферроцена). Комплекс получился из карбонила вольфрама действием метиллития, с последующим алкилированием по атому кислорода. Комплекс оказался удивительно стабильным и очень интересным с химической точки зрения.

Э.О.Фишер
(1918-2007).
Далее были получены многочисленные аналоги этого комплекса, в основном из карбонилов 6-й группы - хрома, молибдена, вольфрама, хотя есть и аналогичные комплексы в других группах. Металл в этих комплексах (металлов 6-й группы) имеет степень окисления 0, а значит способен к эфффективному back-donation. Но у этих комплексов есть две интереснейшие особенности - обязательно один мезомерный донор на "карбеновом" углероде, чаще всего кислород, но также и азот или сера. Но только один! Если таких донора два, то это уже нечто совсем другое. И второе - металл низковалентный, но обязательно имеющий хорошие пи-акцепторные лиганды, лучше всего карбонил. Это "успокаивает" back-donation и сохраняет очень высокую электрофильность карбенового центра.
Для адекватного отображения электронной структуры обычно используют мезомерию и рисуют три граничные структуры. Лиганд относится к типу L и не вносит вклад в степень окисления металла, которая для самых распространённых комплексов этого типа равна 0. Поэтому структурный тип комплекса не отличается от типа самих карбонилов, это координационно-насыщенные комплексы, и это определяет их многие свойства: они устойчивы, а так как все лиганды сидят довольно прочно, для этих комплексов не свойственны реакции с потерей или заменой лигандов, хотя иногда один или два карбонила могут диссоциировать, освобождая координационные места, например, для олефина или ацетилена.
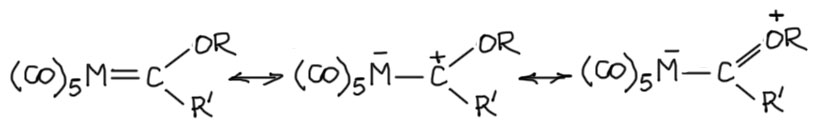
Эффект back-donation играет огромную роль в химии этих комплексов. Большой вклад этого эффекта принято выражать "двойной" связью между металлом и карбеновым центром, но нужно понимать, что этот эффект имеет переменную величину и никогда не достигает такой степени смещения электронной плотности как в карбенах Шрока (об этом дальше). Как и в свободном карбене, у карбенового лиганда в карбенах Фишера есть p-орбиталь, изначально вакантная, но находящаяся под донорными эффектами как со стороны металла через back-donation (левая граничная структура) так и со стороны мезомерного донора, например, алкокси-группы (правая граничная структура). Средняя структура, которую часто называют илидной по причине соседства двух атомов с формальными противоположными зарядами очень удобно показывает электрофильный характер атома углерода, его близость к стабилизированным карбокатионам, или же к карбонильному углероду, наоборот активированному. Чаще всего используется аналогия с производными карбоновых кислот, и тогда металл парадоксальным образом становится аналогом карбонильного кислорода.
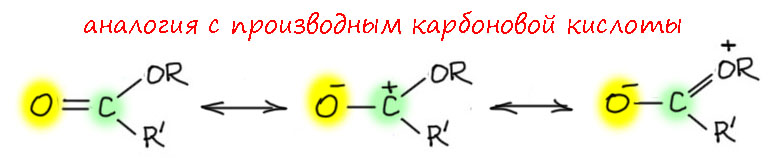
У карбенов Фишера немало реакций, сильно смахивающих на реакции обычных карбонильных соединений, электрофильным центром является карбеновый углерод. Так, они легко реагируют с литийорганикой, образуя аналоги тетраэдрических аддуктов в химии карбонильных соединений, и это очень часто совершенно стабильные комплексы, которые можно заставить отпустить алкоксильную группу только с помощью кислотного катализа. В этом случае получаются новые карбеновые комплексы, которые больше не являются карбенами Фишера, но могут использоваться в разных интересных реакциях, например, они очень пригодились в ранних попытках исследовать механизма реакции метатезиса. Первым эти карбены получил сам Фишер.
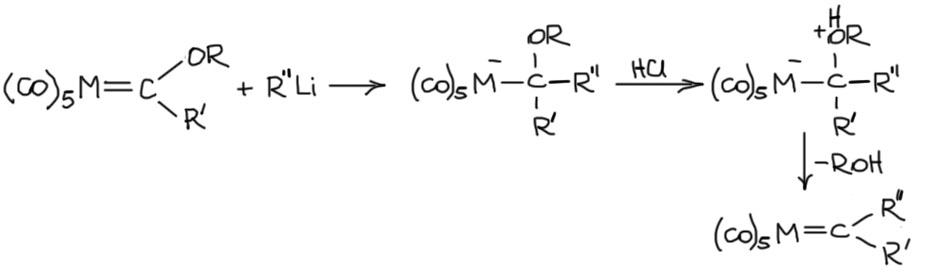
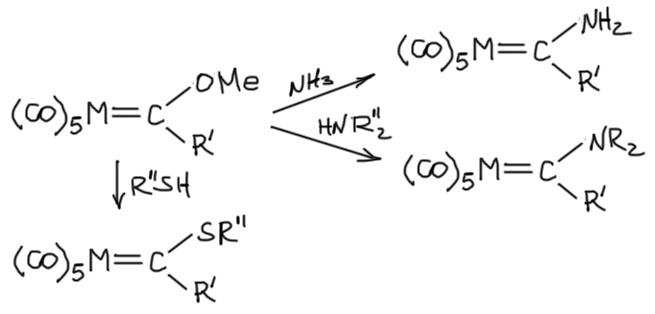
Не менее легко карбены Фишера реагируют с гетероатомными, азотными или серными нуклеофилами, и эти реакции идут просто практически идентично нуклеофильному замещению в производных карбоновых кислот типа переамидирования или переэтерификации. Образующиеся комплексы - типичные карбены Фишера по структуре, свойствам и реакциям, хотя применяют их реже чем их алкоксильные аналоги.
Аналогия с карбонильными соединениями распространяется и дальше - и при наличии атомов водорода на соседнем с карбеновым углероде можно легко получать аналоги енолятов, как в равновесном, так и в стехиометрическом режиме, причём кислотность таких атомов явно существенно выше, чем у обычных карбонильных соединений, и оценивается величинами более близкими к рК малонового эфира или даже нитрометана. У таких енолятов есть все приличествующие енолятам реакции, и конечно есть аналог альдольной конденсации. Из-за высокой кислотности, нуклеофильность таких "енолятов" невелика, и карбонильную компоненту приходится дополнительно раскачивать кислотами Льюиса.
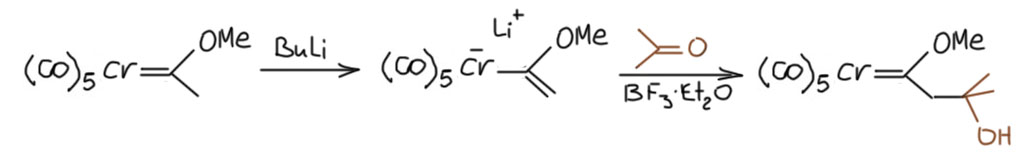
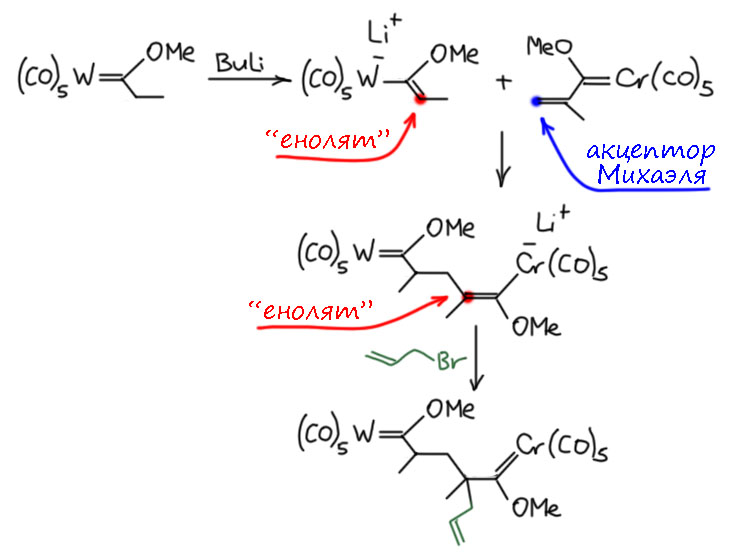
Ну и конечно есть и реакция Михаэля, причём в обоих вариантах - карбены Фишера с двойной связью рядом с карбеновым углеродом (их можно получать, например, дегидратацией "альдолей") работают как отличные акцепторы Михаэля. Можно даже сделать вариант, когда в роли обоих реагентов выступают карбены Фишера, и, например, получить довольно прикольные двойные карбены с разными металлами, скорее всего нафиг никому не нужные, но интересные для демонстрации реакционной способности. Обратим ещё внимание, что при присоединении карбаниона образуется новый "енолят" который можно перехватить ещё одним электрофилом.
Из обычной органической химии хорошо известно такое явление как карбеноиды. Карбены - очень мощная вещь, но, к сожалению, почти совершенно неуправляемая. По утверждению одного из основоположников карбеновой химии, карбен - абсолютный чемпион по неселективности, реагирует почти со всем, что может встретиться в молекулах. Поэтому с самого начала органика искала нечто похожее по типу реакционной способности, но более разборчивое. Поиск не был долгим - уже через несколько лет после появления самого термина карбен, Симмонс и Смит предложили ставший знаменитый реагент, который отлично справлялся с главной задачей карбенов - циклопропанированием олефинов.
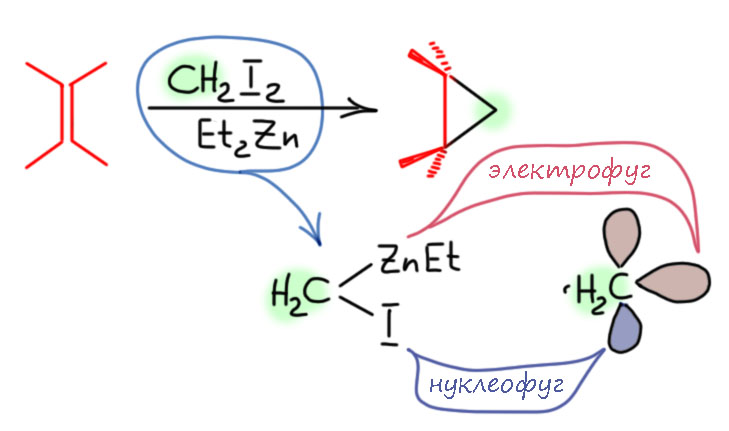
Карбеноиды, основанные на непереходных металлах - цинке, литии, магнии и т.п. - появившиеся в огромных количествах с тех пор эксплуатируют один структурный мотив: наличие на одном атоме углерода двух уходящих групп, одна уходит со своей парой (нуклеофуг, обычно это или галоген или сульфонат), и другая оставляет связевую пару углероду (электрофуг, это всегда металл, возможно с дополнительными лигандами). Эти две уходящие группы формально представляют две орбитали карбена - электрофильную свободную р-орбиталь и нуклеофильную гидридную пару. Точный механизм реакции карбеноидов не всегда хорошо известен, но точно не включает обазования собственно свободного карбена, а скорее является чем-то согласованным, более похожим на реакции активной непереходной металлоорганики.
Но химия карбеноидов парадоксальным образом родилась намного раньше официального дня рождения. С этой химией неразрывно связано такое явление как катализ разложения диазоалканов переходными металлами и их комплексами, а это явление было открыто Людвигом Вольфом в самом начале 20-го века - и мы отлично знаем эту реакцию по методу Арндта-Эйстерта, в котором диазоалканы разлагают разными простыми комплексами серебра. Позже было обнаружено, что разложение диазоалканов вызывают также комплексы меди, и это отличный метод циклопропанирования. За медью пошёл палладий и другие переходные металлы. Возник вопрос - а там что образуется, неужели комплексы карбенов, и тогда такие комплексы карбенов становятся совершенно зачётными карбеноидами. Началось кропотливое изучение этой проблемы, принесшее немалые плоды.
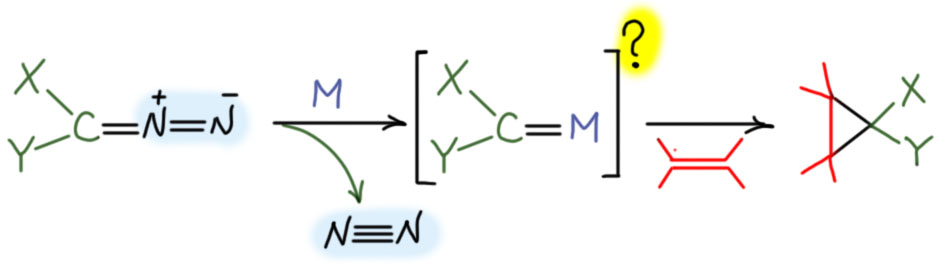
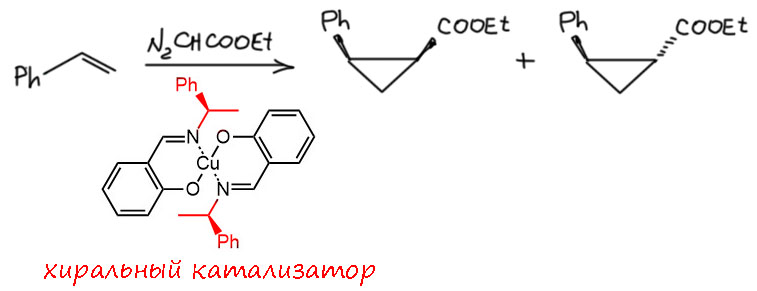
Очень быстро выяснилось, что в такой реакции есть и дополнительный бонус - её можно сделать диастереоселективной и даже энантиоселективной, если взять хиральный комплекс металла. Но поначалу, как обычно, стереоселективность была невелика и до действительно впечатляющих результатов удалось добраться, только тщательно изучив механизм реакции и структуру комплексов.
Хотя комплексов, способных вызывать разложение диазоалканов, огромное количество и самих переходных металлов в этих комплексах немало, одним из важнейших открытий в этой области считается работа группы бельгийских химиков 1976 года, в которой нашли, что димерный ацетат родия работает особенно хорошо, быстренько и чистенько, в каталитических количествах. Интересно, что та же группа чуть раньше предложила для той же цели ацетат палладия, поэтому в далекой от этой области литературе эти два реагента путают, считая двумя равноценными вариантами. Это не так, ацетат родия в этой реакции намного эффективнее, а его необычная структура оказалась важнейшим фактором, определяющим эту эффективность.
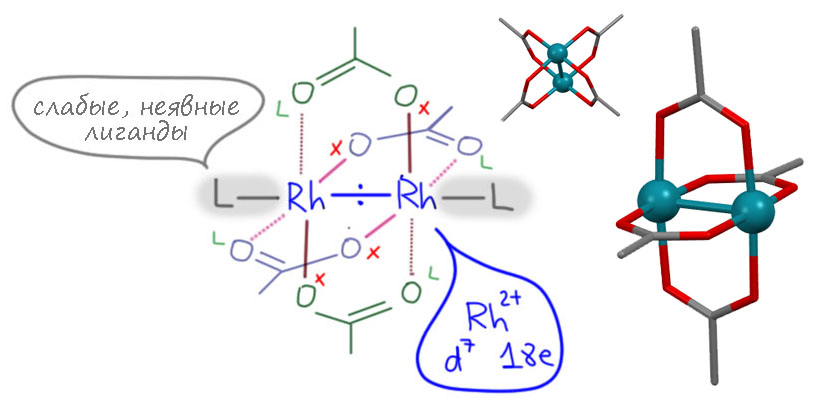
Димерный ацетат родия представляет собой пример комплексов с двухядерным диметальным кластером,атомы металла в которых помимо связи или связей металл-металл, дополнительно скреплены мостиковыми карбоксилатными лигандами. Такие кластеры впервые обнаружил и описал Коттон, причем оказалось, что такой структурный мотив очень распространён среди карбоксилатных комплексов самых разных металлов, и просто невероятно устойчив, сохраняясь в реакциях в неизменности. Лиганды в таких комплексах и сами комплексы метафорически называют "paddlewheel", и отсылают к форме гребного колеса в старинных пароходиках и сполне современных курортных водных велосипедах.
У атома родия в таких комплексах есть по одному месту для внешних лигандов, и там обычно сидит что-то слабосвязанное типа растворителя - это место готово для связывания чего-то посерьёзнее.
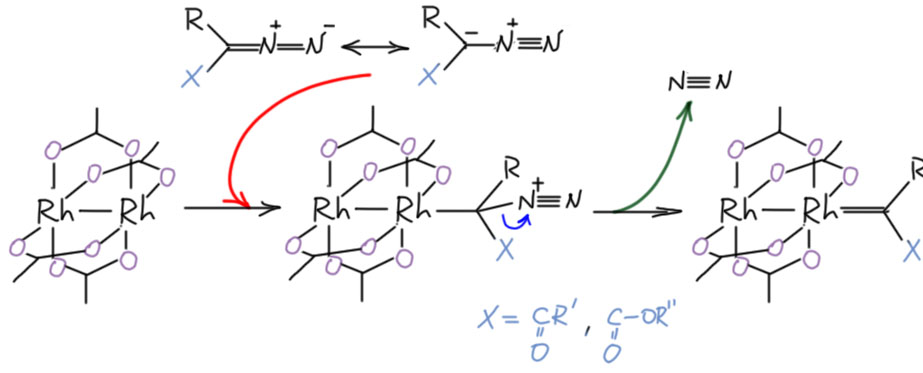
Реакция димерных родиевых карбоксилатов с диазосоединениями протекает очень легко - скоростьопределяющей стадией является замещение молекулы азота на родиевый кластер. Возникает комплекс, в котором есть трёхатомный фрагмент родий-родий-углерод, связь в котором считается трёхцентровой четырёхэлектронной - в таком фрагменте back-donation с атома родия в значительной степени подавлен, и углерод сохраняет практически свободную p-орбиталь, что и обеспечивает высочайшую электрофильность.
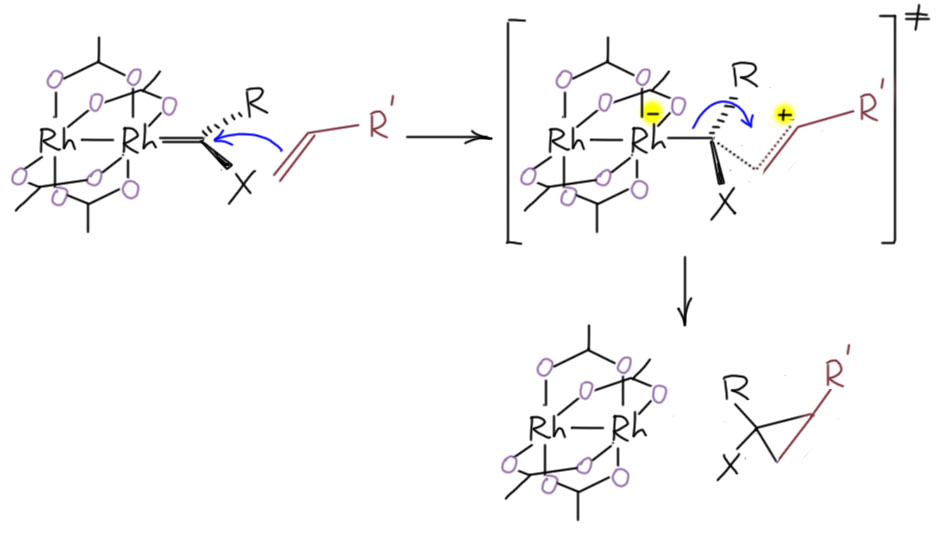
Дальше реакция "летит" - развивается как согласованный процесс взаимодействия с двойной связью олефина, в которой родиевый кластер играет роль электрофугной уходящей группы. Расчеты показывают, что вся реакция развивается с практичеки нулевыми барьерами активации, а сближение карбенового центра и олефина происходит с высокой степенью определённости взаимной ориентации - а это и есть залог высокой стереоселективности.
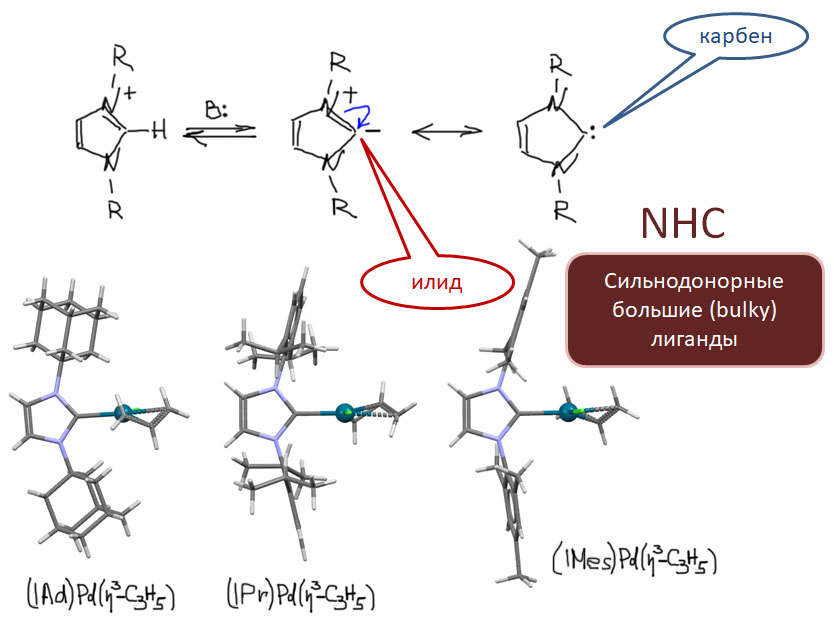
Если на карбеновом углероде ещё более донорные заместители, мы получаем так называемые гетероциклические карбены, открытые в 1960-е Ванцликом и исследованные 30 лет спустя Ардуэнго с сотр. Это как раз вполне устойчивые в свободном состоянии соединения, легко получаемые при депротонировании солей имидазолия и многих других гетероциклов. Эти соединения проявляют признаки и нуклеофильных карбенов и карбанионов. Комплексы таких карбенов (их принято сокращать как NHC - N-heterocyclic carbenes) стали очень популярны в 21 веке, когда понадобились сильнодонорные объёмистые лиганды в разных каталитических процессах.
Отличительная черта этих карбеновых лигандов - высокая донорность и практически полное отсутствие π-кислотности, они не принимают электроны от металла по back-donation. Поэтому эти лиганды являются сильными донорами, очень хорошо стабилизируют высокие степени окисления металлов, а в низковалентных формах делают металл сильнодонорным, нуклеофильным. Особенно популярны такие лиганды с очень объемистыми заместителями - 2,6-дизопропилфенилом, мезитилом, иногда даже 1-адамантилом - такие лиганды успешно заменяют объемистые донорные фосфины, но гораздо дешевле и удобнее их
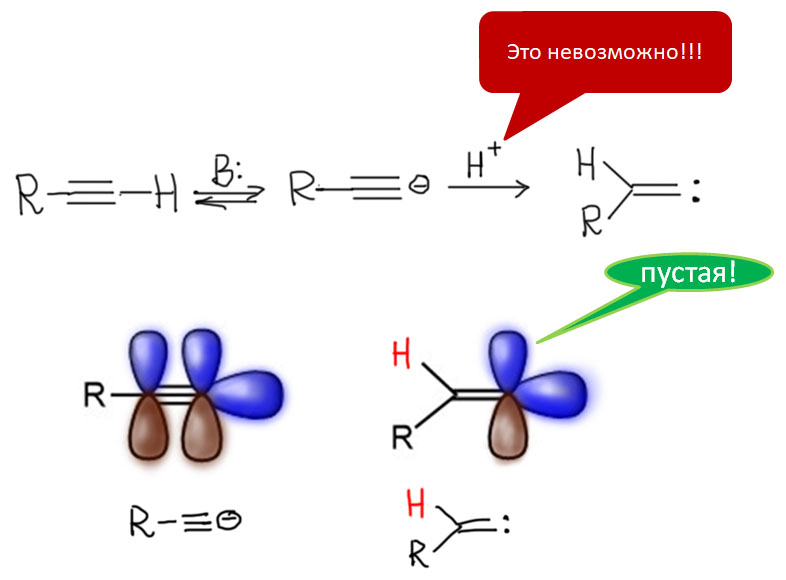
Очень интересный карбен можно получить, по крайней мере, мысленно таким воображаемым экспериментом. Возьмем терминальный ацетилен, оторвем от него протон, и вместо того чтобы положить его на место в обычном кислотно-основном равновесии, возьмем, да и присобачим его к второму атому углерода. Но это же невозможно, скажет любой химик, знакомый с законами кислотно-основных равновесий. Протон всегда садится на место наибольшей основности, точнее, садится в той степени, в которой константа основности превосходит константы основности по другим основным центрам молекулы. Но мысленно нам никто не запрещает приделать его так. Одна из пи-связей ацетилена при этом уйдет на образование связи C-H, а на атоме с неподеленной парой останется пустая p-орбиталь. Все признаки карбена - винилидена - налицо. Жаль только, что сделать этого реально нельзя, а то мы получили бы очень интересный изомер ацетилена. Но когда в дело вступают переходные металлы, экзотическая и скорее воображаемая молекула внезапно становится важнейшим игроком.
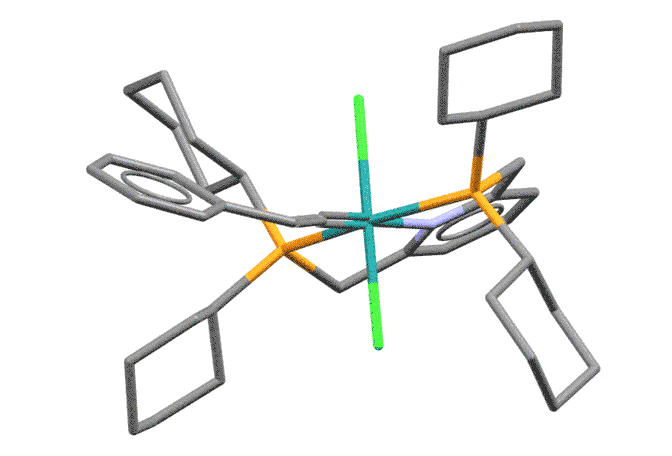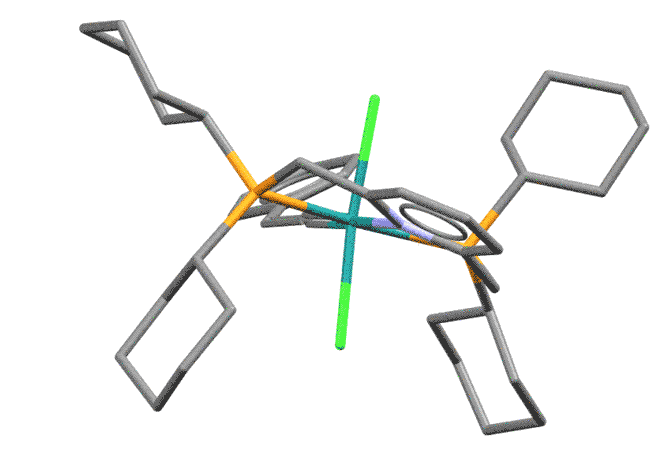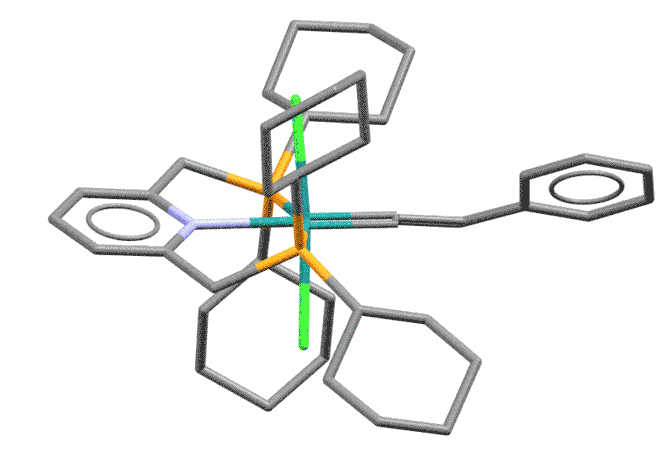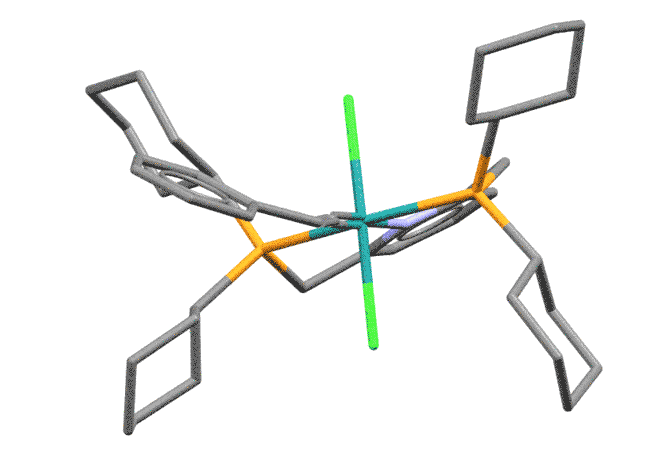
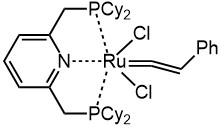
Пример винилиденового комплекса Ru(2+)
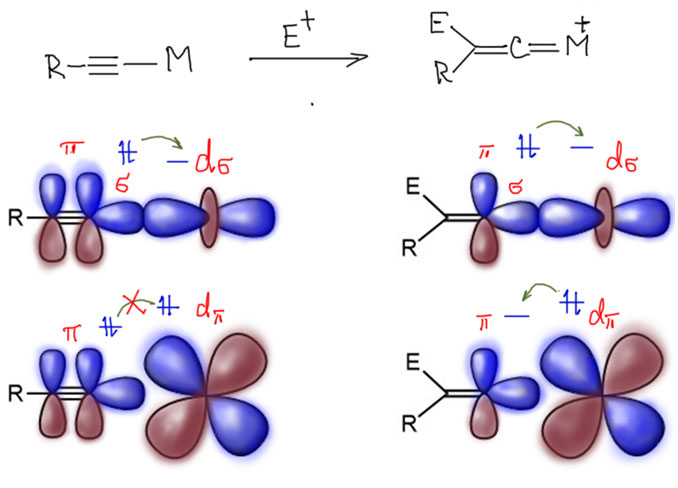
В отличие от свободного винилидена, комплексы винилиденов хорошо известны для многих переходных металлов. Они образуются при атаке электрофилов на ацетиленовые комплексы. После атаки электрофила бывшая π-связь как раз и превращается в p-орбиталь карбенового атома, а обратный донорный эффект, включающийся после такого перераспределения электронной плотности, как раз стабилизирует это состояние, притом что в исходном ацетилениде те же орбитали были заселены парами электронов каждая, то есть их взаимодействие было дестабилизирующим, отталкивательным. Выгодность образования винилиденовых комплексов - одна из причин весьма широкого вовлечения винилиденовых комплексах в реакции терминальных ацетиленов с участием поздних переходных металлов.
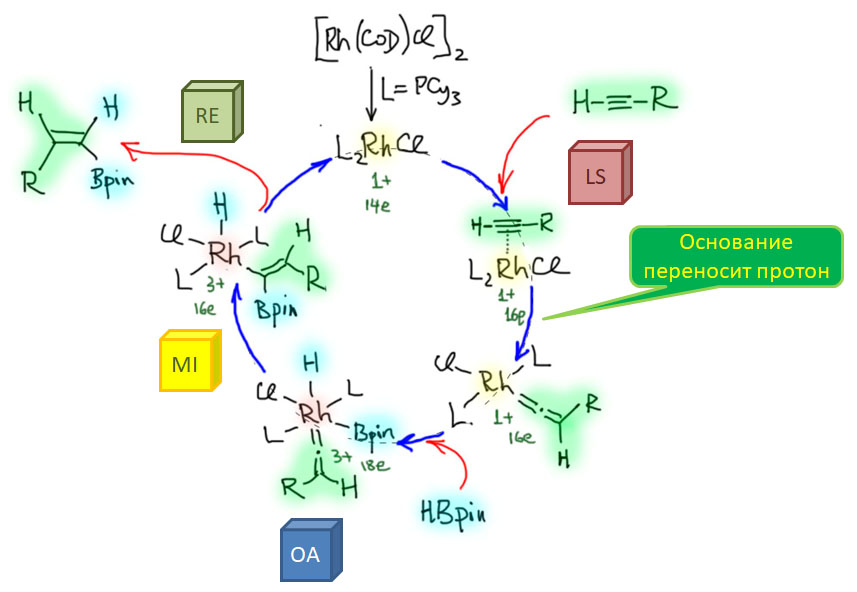
Одно из интересных проявлений образования винилиденовых комплексов - каталитическое транс-гидроборирование, обнаруженное, как и почти все остальное, касающееся всех и всяческих методов каталитического борилирования, Мияурой с сотр. (J. Am. Chem. Soc. 2000, 122, 4990-4991) . Как мы знаем, комплексы родия, например, катализатор Уилкинсона катализируют реакцию гидроборирования, но стереохимия этой реакции остается обычной - син-присоединение борана. Но в присутствии донорного фосфина и основания, стереохимия драматически изменяется. Необычная стереоселективность и еще более необычное перемещение протона с терминального конца ацетилена на β-атом углерода, подтвержденное дейтериевой меткой, как раз и являются следствием участия такого интермедиата в каталитическом цикле.
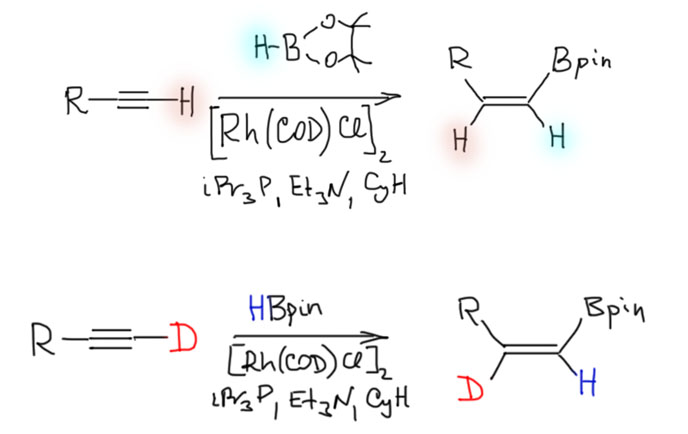
Каталитический цикл транс-гидроборирования включает образование винилиденового комплекса, причем протон от терминального углерода ацетилена переносится на второй атом. Далее следует окислительное присоединение борана, миграционное внедрение карбена в связь металл-бор, и восстановительное элиминирование продукта. Самый важный вопрос - почему получается именно транс-продукт, скорее всего объясняется стерическим отталкиванием металла и группы R ацетилена. В согласованных механизмах, а миграционное внедрение - типичная согласованная реакция, - стерические факторы всегда являются очень важными и часто определяющими.
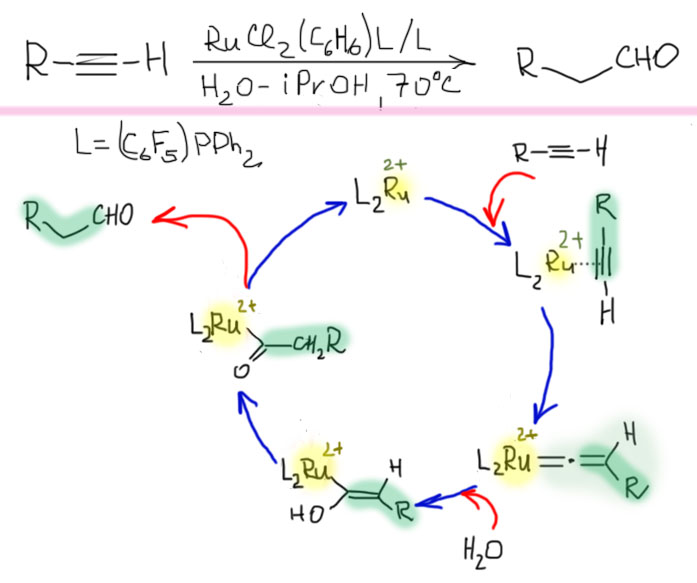
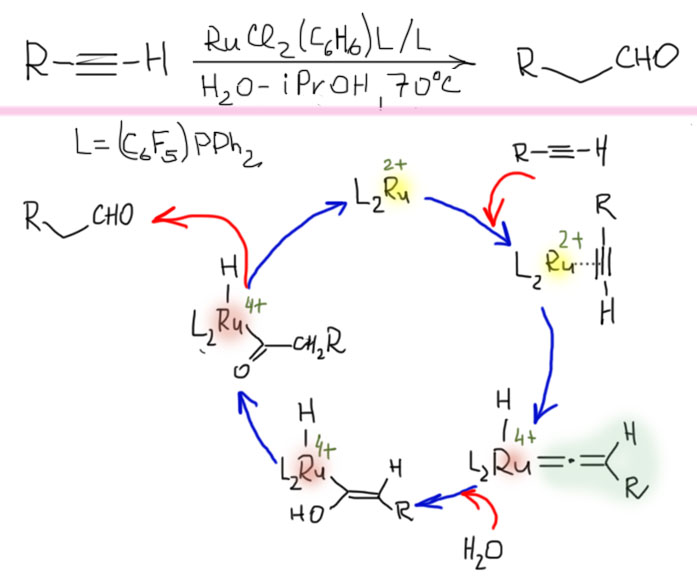
Винилиденовые комплексы вовлечены и в другие аномальные реакции ацетиленов, например, присоединение нуклеофилов к терминальному атому углерода (например, гидратация против правила Марковникова, анти-марковниковское присоединение). Эта необычная реакция обнаружена Вакацуки и сотр. (Angew. Chem. Int. Ed. 1998, 37, 2867), и позже получила развитие в множестве работ по присоединению и других нуклеофилов. Для реакции требуются фосфиновые лиганды с повышенной пи-кислотностью, обусловливающие электрофильность атома углерода рядом с металлом в винилиденовом комплексе. Присоединение нуклеофила (воды) дает ацильный комплекс, и далее связь с металлом разрывается за счет протолиза.
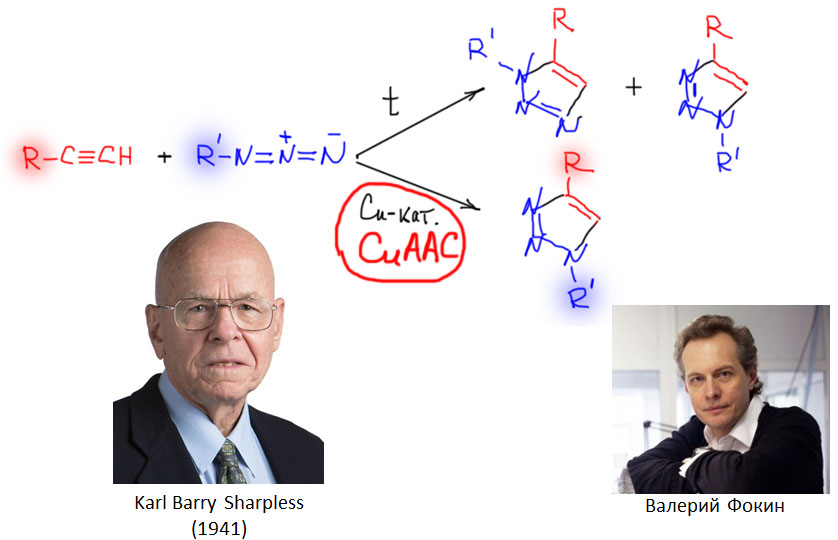
Самая знаменитая реакция, обусловленная участием винилиденовых комплексов, - так называемая клик-реакция. Понятие "клик-реакции" предложил в 2001 нобелевский лауреат (не за это) Карл Барри Шарплесс, определив критерии, которым должна удовлетворять реакция (точнее, метод), чтобы иметь право так называться: универсальность, высокие выходы продуктов и слабая зависимость выходов от структуры реагентов, высокая селективность, простота исполнения, и т.п. Первую реакцию, удовлетворяющую этим критериям он же, в сотрудничестве с В.Фокиным, и описал. Эта реакция формально является хорошо известным диполярным [2+3]-циклоприсоединением, известным как реакция Хьюсгена. Но это сходство формально. Некатализируемая реакция Хьюсгена - согласованная реакция, по механизму похожая на еще более знаменитое [2+4]-циклоприсоединение Дильса-Альдера. Реакция Хьюсгена очень сильно зависит от структуры субстратов, часто идет очень медленно, с низкой селективностью. Клик-реакция Фокина-Шарплесса, получившая сокращенное обозначение CuAAC (Cu catalyzed azide-acetylene condensation) идет в присутствии очень простого Cu катализатора, и отличается очень высокой селективностью, выходами, и прочими достоиствами. Реакция стала невероятно популярна как универсальное средство соединения фрагментов с разной функциональностью в большие многофункциональные молекулы.
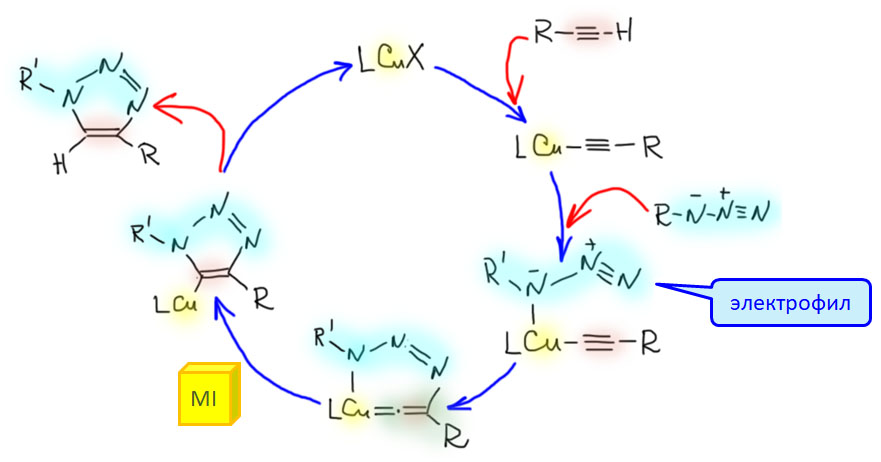
Для объяснения CuAAC-клик реакции используется несколько механизмов, общей чертой которых является участие винилиденового комплекса меди. Приведем более простой механизм с участием одного атома меди на каталитический цикл (есть еще двух-медный механизм). Ацетиленид меди подбирает азид в виде L-лиганда. Крайний атом азота в молекулах азидов обладает слабой электрофильностью (это очень похоже по структуре на соль диазония, известную из обычной органической химии, и также являющуюся слабым азотным электрофилом). Электрофильный азот внутримолекулярно садится на дальний углерод ацетиленида, что и дает винилиденовый комплекс. Далее следует обычное миграционное внедрение карбена по связи металл-азот, и протолиз связи углерод-медь. Для такого каталитического цикла нужна Cu(1+), но не требуются никакие специфические лиганды. Cu(1+) обычно генерируют прямо в реакционной смеси (пред-активация) из предкатализатора - банального сульфата меди(II), восстановлением натриевой солью аскорбиновой кислоты (витамином C). Реакция идет при комнатной температуре или небольшом нагревании. Такой механизм реакции однозначно объясняет, почему образуются только 1,4-дизамещенные 1,2,3-триазолы в реакции азида и ацетилена "голова-к-хвосту", но не образуется обычный в некаталитической реакции Хьюсгена изомер "голова-к-голове" (почему-то никогда не говорят "хвост-к-хвосту", хотя это точнее и образнее).
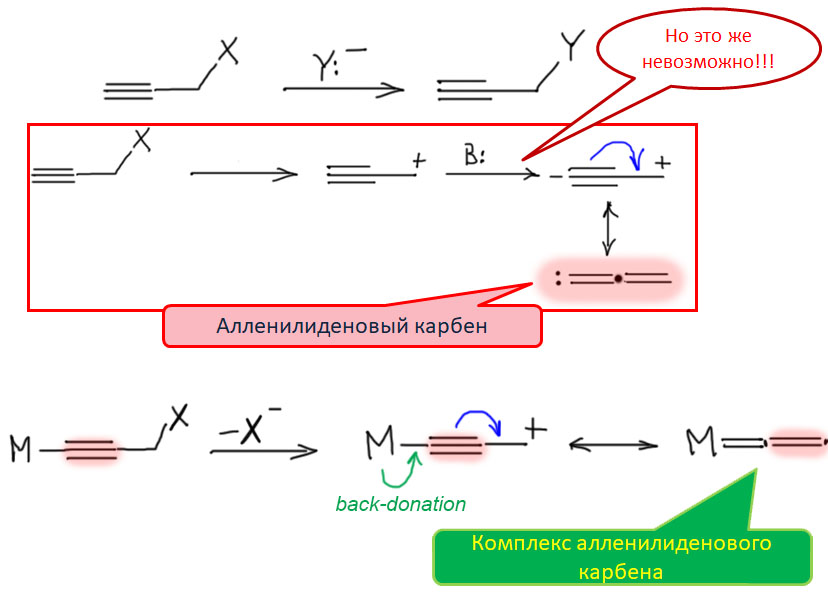
Та же логика, что лежит в основе химии комплексов винилиденовых карбенов, может привести еще дальше - к карбену с кумулированной системой двойных связей алленового типа - алленилидену. Свободный карбен такого типа можно представить себе, как результат депротонирования пропаргильного катиона, но такой процесс кажется невозможным (возможно, зря, но это другой вопрос). Если же то же самое происходит в пропаргильной системе, связанной в виде ацетиленидного комплекса с металлом, то мы получим аленилиденовый карбен в комплексе.
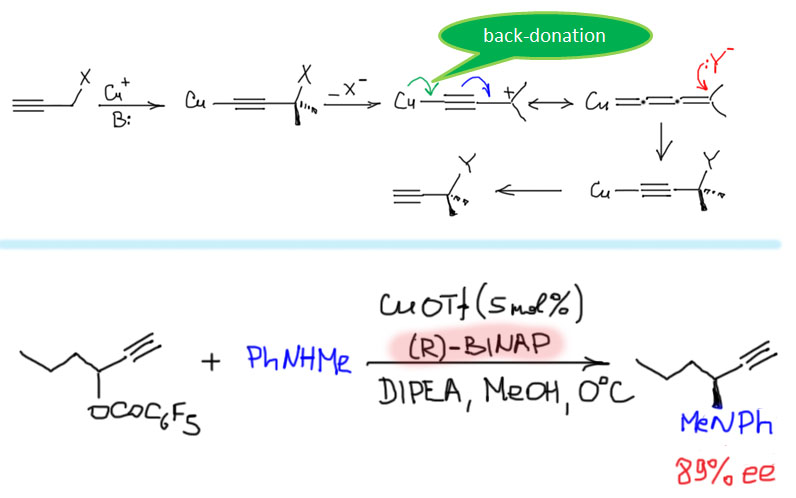
Алленилиденовые комплексы, как и винилиденовые, оказались весьма полезными интермедиатами в катализе. Одна из реакций, где их участие доказано, это пропаргильное замещение в присутствии комплексов меди и других переходных металлов. В обычной органической химии, пропаргильное замещение очень распространено, и является просто аналогом аллильного замещения. В химии переходных металлов это не так, и прямого аналога каталитического аллильного замещение нет, просто потому что пропаргильная система не дает аналогичных аллильных тригапто-комплексов: линейная геометрия пропаргильного фрагмента препятствует такому типу связывания. Поэтому в течение долгого времени каталитическое пропаргильное замещение оставалось нерешенной проблемой. Менее 10 лет назад такие процессы появились, и стали быстро развиваться, прежде всего в энантиоселективном варианте. На слайде приведен типичный пример такой реакции в присутствии комплекса меди с (R)-BINAP из рацемических пропаргильных эфиров образуются оптически активные амины (A. Yoshida et al, Org.Lett., 2011, 13, 2460–2463).
Карбеновых комплексов переходных металлов, как видим, очень много, и реакций с их участием тоже немало. Но всё-таки самый важный процесс с участием карбеновых комплексов переходных металлов – метатезис. Эта реакция имеет огромное значение и для промышленного крупнотоннажного синтеза, и для тонкого органического синтеза. Метатезис вполне заслуживал бы отдельной лекции, если бы у нас было на это время. Поэтому пока ограничимся кратким обзором ее особенностей и возможностей.
Метатезис, то есть просто обмен, это любая реакция в которой два реагента меняются друг с другом частями. Это очень важная реакция, точнее, целое семейство реакций и в тонком синтезе, и в промышленном. За исследование и развитие метатезиса в 2005 году была присуждена нобелевская премия.
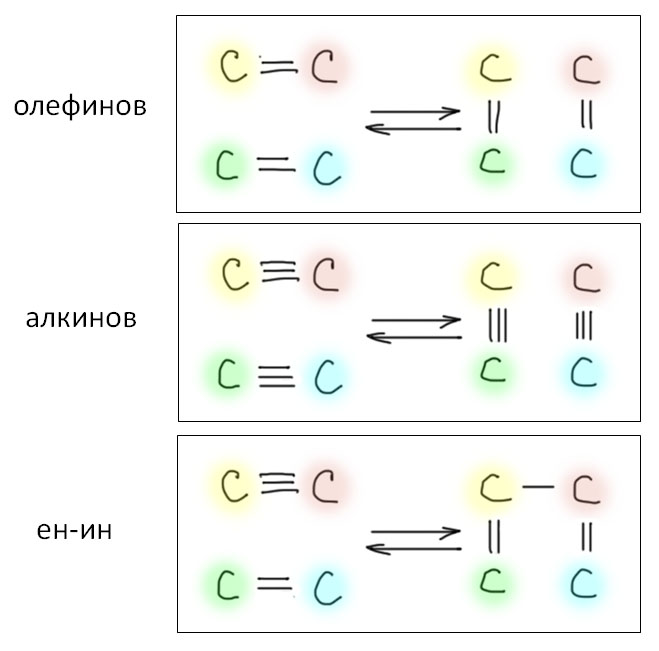
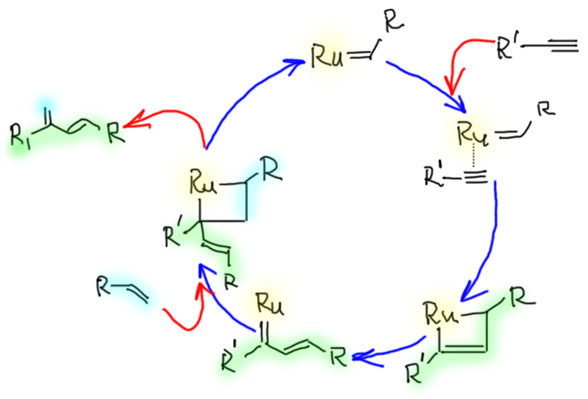
Наиболее хорошо известен и изучен метатезис олефинов, в котором происходит равновесный обмен половинками – карбенами.
Метатезис ацетиленов также очень неплохо изучен – в нем ацетилены размениваются своими половинками, карбинами.
В паре олефин – ацетилен фактически происходит именно олефиновый метатезис, поэтому реагент с тройной связью сохраняет целостность: в реакции ен-инового метатезиса образуются сопряженные диены.
Реакция метатезиса была известна с 1960-х как побочная реакция в полимеризации олефинов, катализируемой комплексами ранних переходных металлов (катализаторы Циглера-Натты). Развитию собственно метатезиса долго мешало отсутствие понимания того, как происходит эта, на первый взгляд, совершенно невероятная реакция. Механизм реакции метатезиса предложил в 1970 французский исследователь Ив Шовен (нобелевский лауреат 2005 г, J.L.Herisson, Y.Chauvin, Makromolekulare Chemie 141 (1970) 161-176). Была установлена ключевая роль карбеновых комплексов металлов, а собственно метатезис происходит в последовательности реакций [2+2]-циклоприсоединения и ретро-[2+2]-циклоприсоединения, в котоых участвуют четырёхчленные металлациклы. Все реакции цикла обратимы.
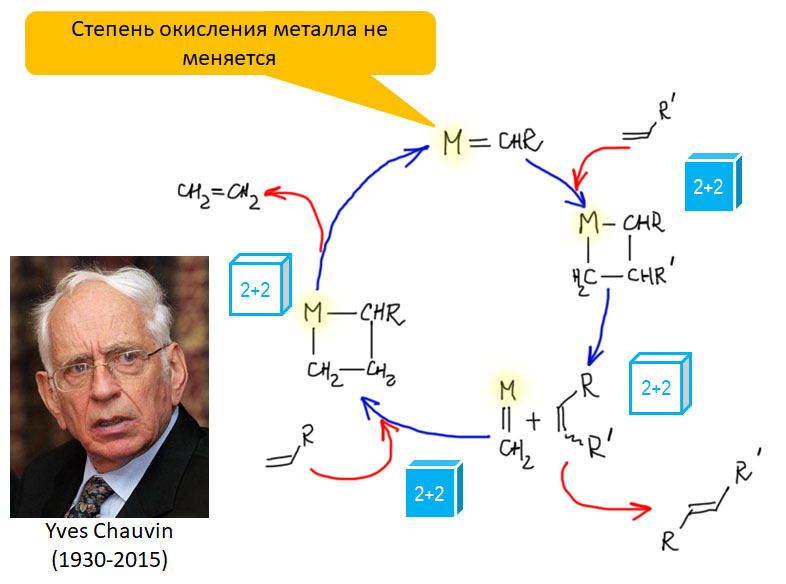
В механизме Шовена оставалось неизвестным, как образуется первый карбеновый комплекс, то есть как происходит пред-активация пред-катализаторов, в роли которых в ранний период истории этой реакции служили разнообразные оксида, соли и комплексы металлов от 4-й до 8-й групп. В этот период реакция метатезиса была бесполезна для органического синтеза, так как всегда давала смеси олефинов, а удобных катализаторов для неё придумать не могли. Механизм Шовена дал ключ для более целенаправленной работы над дизайном эффективных катализаторов, и в эту работу включилось несколько очень амбициозных исследователей. Запахло успехом.
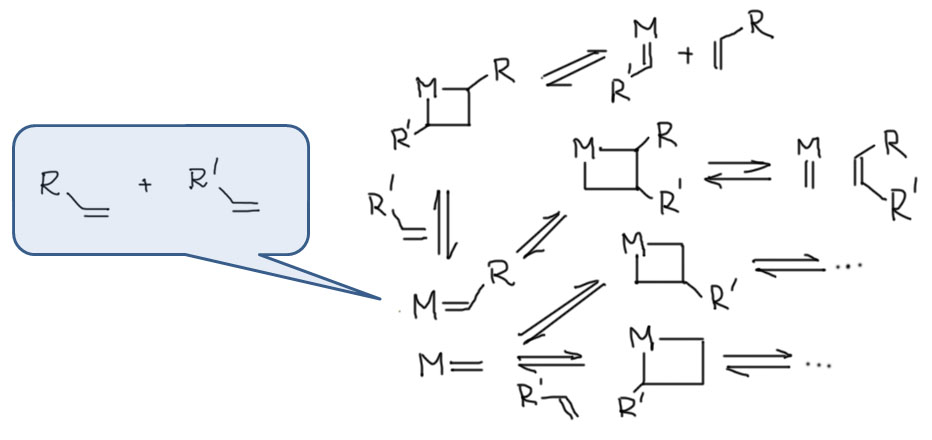
Эта проблема заложена в самом механизме процесса - его обратимости и весьма низкой чувствительности к структуре исходных олефинов. Поэтому в произвольном случае кросс-метатезис (реакция двух разных олефинов, даже монозамещенных) не может дать один продукт, даже если не учитывать стереоселективность (цис-транс, например), которая также далека от желаемой в современном синтезе. Эти врожденные особенности обусловлены природой механизма и обратимостью, и не могут быть преодолены в общем случае подбором катализатора или условий, хотя в ряде частных случаев, когда реакцию удается переключить в режим кинетического контроля, удается достичь селективности. Все стадии процесса обратимы, а следовательно и весь процесс обратим, и все возможные олефины находятся в равновесной смеси, состав которой определяется или относительной устойчивостью олефинов (при установившемся равновесии), или принципом Ле Шателье, если выполняются условия применения этого принципа (избыток одного из исходных олефинов, уход одного из продуктов из реакционной смеси). Безусловно, далеко не все равновесия равновероятны, и в конкретных случаях выделяются основные направления процесса,
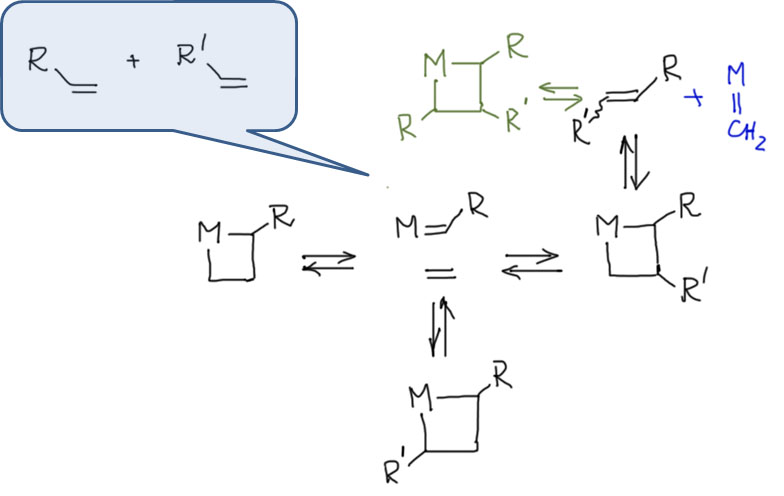
Из механизма Шовена стало ясно, что катализаторами метатезиса должны быть карбеновые комплексы переходных металлов, скоре всего, ранних. Такие катализаторы впервые нашёл Ричард Шрок среди специфических карбеновых комплексов, которые сильно отличаются от известных в то время карбенов Фишера. В таких комплексах карбеновый лиганд (и аналогичные нитреновый) работают как кислород в тех же триоксидах, то есть как X2-лиганды, дающие вклад +2 в степень окисления металла. Такие карбеновые комплексы (не сами лиганды!) принято называть карбенами Шрока.Эффективные катализаторы метатезиса олефинов используют готовые (pre-formed) комплексы с карбеновыми лигандами. Первые катализаторы этого типа (катализаторы Шрока, Schrock Mo и Schrock W) были построены на основе комплексов ранних переходных металлов в высоковалентном состоянии, изоэлектронные по центральному атому металла с триоксидами Mo(6+) и W(6+).
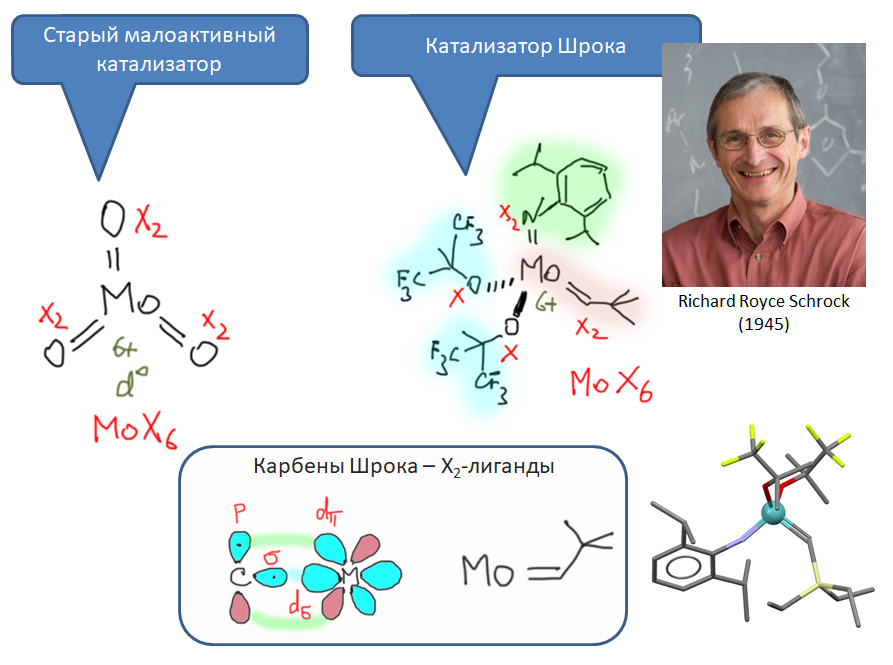
Прежде чем найти применение в метатезисе олефинов один конкретный карбен Шрока проявил себя в знаменитой реакции олефинирования по Теббе, и в её многочисленных вариантах, например, реакции Петасиса, очень популярной в современном органическом синтезе из-за потрясающей мягкости и универсальности. В этой реакции проявилась высокая нуклеофильность карбена Шрока, позволяющая атаковать карбонильный углерод не только альдегидов и кетонов, но даже и сложных эфиров и амидов (X = OR, NRR'). Реакция является аналогом не менее знаменитой реакции Виттига, поэтому ее часто игриво называют металло-Виттигом. А механизм реакции очень сильно напоминает механизм метатезиса, хотя и не является каталитическим так как нет способов превратить оксо-комплекс титана в карбен.
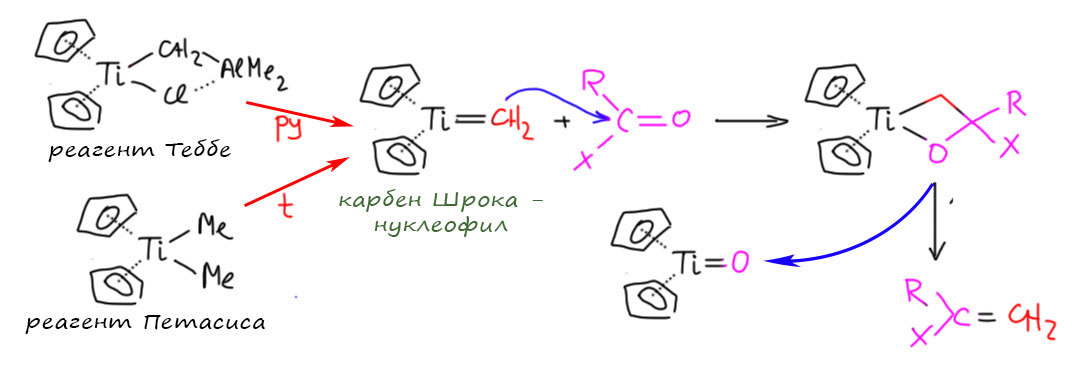
Титановый карбен Шрока получают прямо в реакционной смеси или из реагента Теббе, который представляет собой просто комплекс этого карбенового комплекса с диметилалюминийхлоридом, или из реагента Петасиса, диметилтитаноцена.
В этих реакциях очень ярко проявляется природа карбенов Шрока, которые представляют собой результат полного смещения электронной плотности с раннего переходного металла через back-donation с изменением степени окисления до максимальной и конфигурации до d0.
Катализаторы Шрока решили проблему, как сделать метатезис полезной реакцией в синтезе, но не сделали ее широко популярной, так как были сложны в использовании, и несовместимы с многими типами заместителей в олефинах. Настоящую революцию в метатезисе совершил Роберт Граббс, который предложил очень удобные рутениевые катализаторы, доступные и простые в использовании. После этого метатезис быстро стал одним из основных методов орагнического синтеза, ежегодно используемый в тысячах работ.
В катализаторах Граббса карбены - обычные L-лиганды (и это не карбены Фишера, как часто можно услышать!), весьма простые. В таких комплексах карбены - L-лиганды. Несколько поколений таких комплексов предложил Граббс и его последователи. Замена фосфина карбеновым лигандом и хелатирующим карбеном повышает активность комплексов (TOF) и их долговечность (TON), хотя катализатор 1-го поколения до сих пор широко используется.
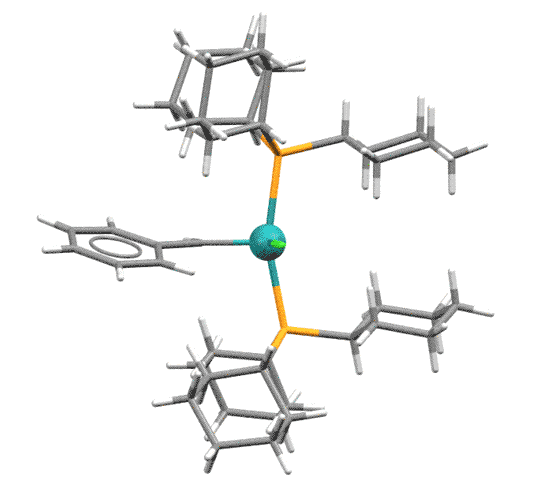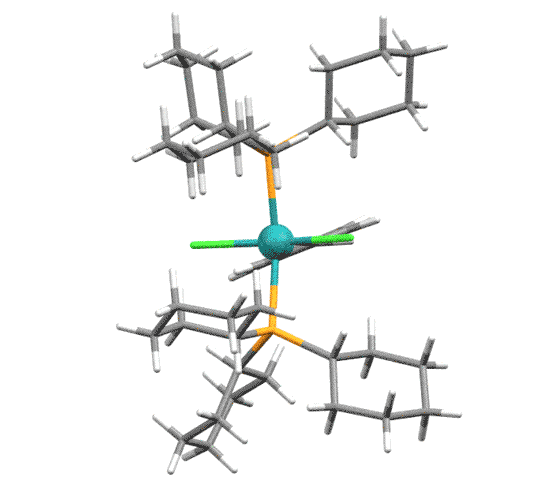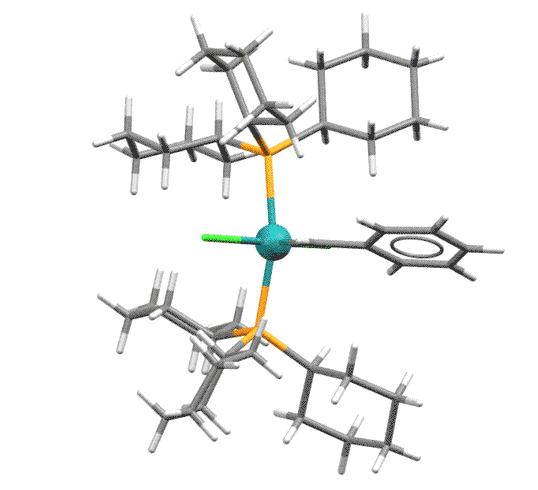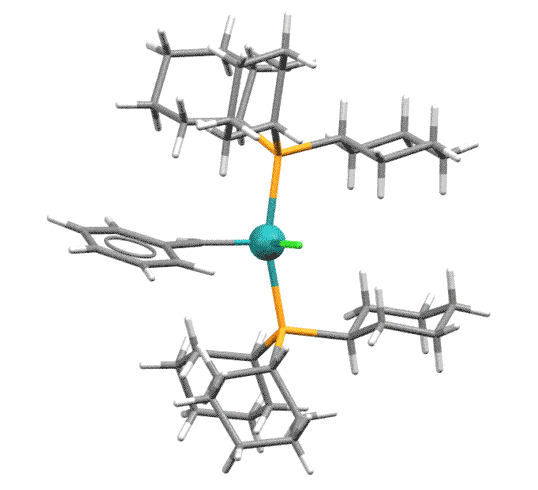
После появления бис-фосфинового катализатора 1-го поколения (Grubbs-I) стало ясно, что фосфин - причина проблем, в частности слабых TON.
В следующих поколениях сначала один фосфин был заменён на NHC-лиганд IMes или SIMes (Grubbs-II ) - обратите внимание что в таком комплексе два карбена разных типов.
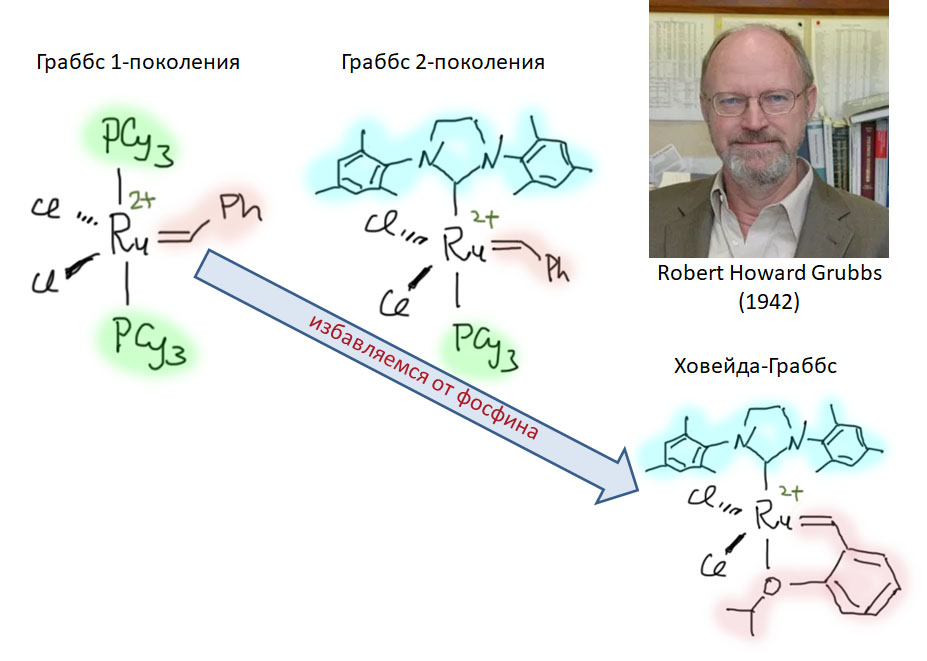
Следующее важное усовершенствование внёс американский химик иранского происхождения Амир Ховейда - он заменил и второй фосфин, причём не на отдельный лиганд, а на карбен с возможностью хелатирования. Катализаторы Ховейды-Граббса 1-го поколения ( с фосфином) и второго поколения (вообще без фосфина, но с NHC) HG-I и HG-2 в современном синтезе особенно популярны, так как, кроме прочего, они дают лучшие результаты в кросс-метатезисе.
Основной вид метатезиса, применяемый в тонком органическом синтезе, - циклизация (Ring Closure Metathesis, RCM), для которой почти всегда берут диены с двойными связями на концах цепи. Метатезис приводит к замыканию цикла и образованию второго олефина - этилена, самопроизвольно уходящего из реакционной смеси, и тем самым смещающего равновесие метатезиса в сторону продукта циклизации.
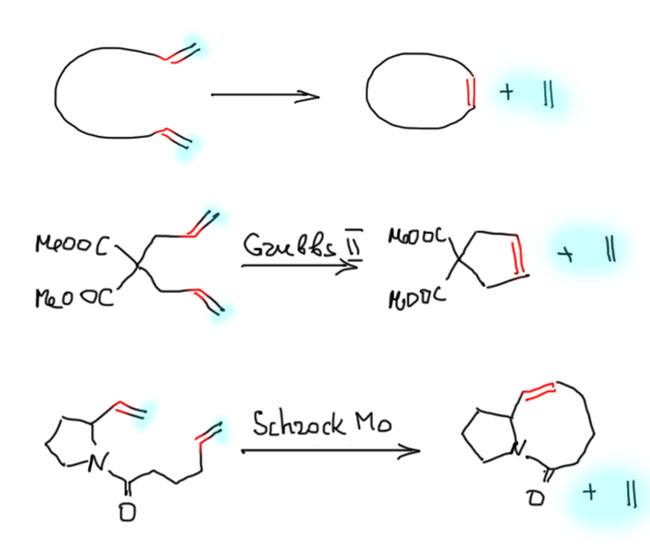
Этот вид метатезиса позволяет получать почти любые циклы, кроме циклопропана и циклобутана, а не только 5- и 6-членные. Метод очень востребован в синтезе больших циклов (макроциклов), хотя и имеет врождённый и почти непреодолимый недостаток - низкая стереоселективность, образование смесей (E)- и (Z)- изомеров, относительно образовавшейся двойной связи. Для реакции годятся оба основных типа катализаторов - и Шрока и Граббса, катализаторы Шрока чаще используют для образования макрогетероциклов. Метод был открыт в 1980 г одновременно Виллеменом и Цудзи ещё до открытия высокоактивных катализаторов Шрока и Граббса, и с тех пор получил значительное развитие и множество новых удобных протоколов. Например, реакция отлично идёт в сверхкритическом CO2.
Метатезис алкинов - более редкий метод. Он построен на тех же принципах, но использует не карбеновые, а карбиновые лиганды. Основные катализаторы принадлежат к типу Шрока, являются производными высоковалентных ранних переходных металлов, молибдена и вольфрама. Карбиновый лиганд в таких комплексах относится к типу X3.
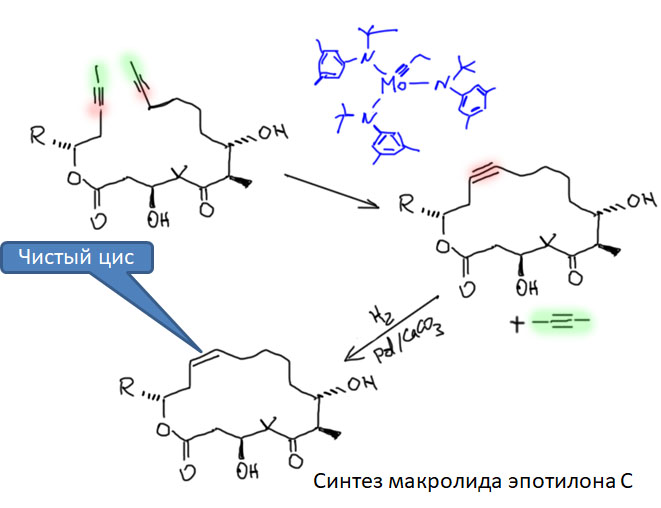
Из-за невозможности образования циклов с тройной связью и числом атомов менее 8, метод применяют только для образования больших циклов, иногда вместо олефинового метатезиса, если необходима точная стереохимическая конфигурация двойной связи (цис или транс). Метатезис олефинов, к сожалению, нестереоспецифичен и часто дает смеси цис- и транс. RCM метатезис алкинов дает циклический алкин, который далее подвергают стереоспецифическому гидрированию. В отличие от RCM метатезиса алкенов, в метатезисе алкинов избегают терминальных ацетиленов, склонных к побочным реакциям, но все равно берут "маленькие" заместители, чтобы второй пробукт метатезиса был легколетуч и быстро удалялся из реакционной смеси. Такой подход эффективно решает проблемы синтеза многих важных макроциклических соединений, в частности, природных макроциклических лактонов (макролидов). В частности, очень важный химиотерапевтический препарат эпотилон имеет цис-конфигурацию двойной связи, и многочисленные попытки разработать синтез этого соединения с помощью алкенового RCM к успеху не привели - получавшиеся смеси практически разделить не получалось. Алкиновый RCM проблему решил.
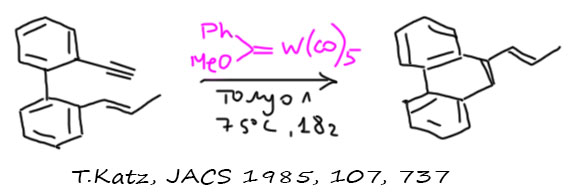
Ещё более нетривиальный вид метатезиса открыл Томас Кац в 1985 году. Используя в качестве катализатора карбен Фишера он показал, что немного авантюрная идея разобрать алкин на двойную и простую связь, использовать первую в метатезисе, а вторую, чтобы соединить две новые двойные связи в один диен, вполне работает. Новая реакция показала, что метатезис является по настоящему общей реакцией.
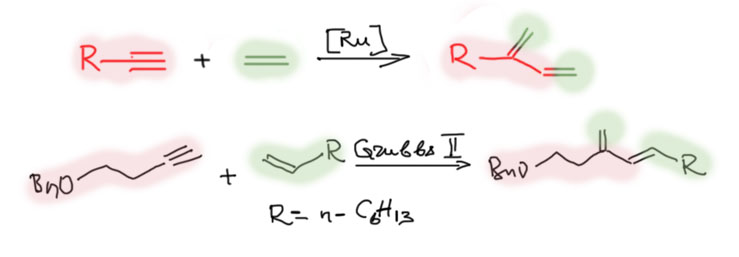
Карбены Фишера действительно являются неплохими катализаторами этого типа метатезиса, но в дальнейшем и в этом методе воцарились новые катализаторы Граббса. Ен-иновый метатезис гораздо сложнее представить, чем ен-еновый и ин-иновый, так как всегда трудно поверить в то, что бывший ацетилен не превращается в экзо-двойную связь, а буквально зарывается внутрь скелета новой молекулы.
Каталитический цикл ен-инового метатезиса ведёт карбен, как в ен-еновом, а не карбин, как в ин-иновом, но цикл включает и металлациклобутен, и металлациклобутан.
Цикл весьма сложен и на каждой стадии предоставляет возможности для альтернативного развития событий. Поэтому реакции ен-инового метатезиса часто бывают совсем неселективны и лечению новыми катализаторами не поддаются, а примеров успешного применения этой реакции на порядки меньше чем его более простых родственников. Тем не менее, когда всё хорошо продумано и получается, результат впечатляет красотой замысла и исполнения.