
Нуклеофильность в химии переходных металлов
Металлы всегда ассоциируются с кислотностью Льюиса. В образовании координационных соединений они предоставляют вакантные орбитали, принимая электроны от лигандов. Поэтому вопрос о нуклеофильности соединений металлов можно было бы считать абсурдным. Но это не так. Атомы металлов во многих комплексах проявляют реакционную способность, очень похожую на традиционное понятие о нуклеофильности и реагируют с органическими электрофилами. Более того, они делают это часто с большим успехом, чем самые нуклеофильные из традиционных органических нуклеофилов. Но проблема эта не так проста – реакции комплексов переходных металлов, которые можно классифицировать как нуклеофильные довольно разнообразны, и можно выделить не один, а несколько механизмов такого действия. Один из них – окислительное присоединение – особенно важен, но мы не можем не посмотреть на другие возможности, хотя бы для того, чтобы получше понять мощь и необычность этого важнейшего механизма, задействованного во множестве процессов с участием комплексов переходных металлов.
Настоящее нуклеофильное замещение – классический механизм SN2
Когда мы изучаем алифатическое нуклеофильное замещение в курсе органической химии, то много внимания уделяем механизму SN2. Это не случайно, SN2 – действительно один из наиболее важных механизмов в органической химии, по этому механизму идут сотни и тысячи важных органических реакций, а раз разобравшись в закономерностях, можно с успехом применять эти знания и управлять реакционной способностью, подбирая нуклеофилы, растворители, уходящие группы, активирующие добавки, разбираться в том, когда этот механизм работает, и продукт замещения может быть получен, – а когда шансов получить продукт замещения нет никаких.
Среди закономерностей SN2 одна из важнейших – зависимость нуклеофильности различных частиц от положения нуклеофильного центра в Периодической системе. Многие помнят, что в группах нуклеофильность изменяется сверху вниз – атомы становятся больше, и связь пары электронов, отвечающей за нуклеофильность, становится слабее. Вообще-то это довольно парадоксальное обобщение: ведь одновременно ослабевают и образующиеся в результате замещения связи, что неизбежно должно было бы ухудшать энергетику процесса замещения – ведь мы теряем энергию связи с уходящей группой, и приобретаем энергию связи с входящей (с поправками на сольватацию, но это не так важно). Получается, что для SN2, для скорости реакции замещения, не так важна энергетика связей, как важна возможность взаимодействия в переходном состоянии, когда связи разорваны/образованы не полностью. И в первый ряд выходит размер электронных облаков неподеленной пары – чем они больше и удаленней от ядра, тем раньше завязывается взаимодействие – образуется так называемое рыхлое переходное состояние. И это отлично работает. Но – только для элементов главных подгрупп – металлов, металлоидов и непереходных металлов. Внизу главных подгрупп мы находим самые сильные нуклеофилы – супернуклеофилы. Супернуклеофилами принято называть такие нуклеофилы, скорость реакций которых с типичным субстратом (обычно берут MeI) оказывается больше, чем скорость смешения реагентов. Вы капаете каплю раствора нуклеофила в раствор субстрата, и реакция проходит раньше, чем капля разойдется по объему колбы, даже если мешалка крутится как ненормальная. Такие скорости невозможно измерить обычными методами химической кинетики, и приходится применять всякие модные ухищрения и навороты. Нам это сейчас не интересно, просто примем к сведению, что существуют такие нуклеофилы, которые нуклеофильнее всех прочих нуклеофилов. И обитают такие нуклеофилы в главных подгруппах Таблицы внизу. Можно сказать по образцу известного сочинения Джорджа Оруэлла: Все нуклеофилы нуклеофильны, но некоторые нуклеофилы нуклеофильнее всех прочих нуклеофилов.

SN2 в химии переходных металлов
Среди комплексов переходных металлов также встречаются нуклеофилы для SN2 на атоме углерода. Как это можно определить? Да, очень просто – с помощью реакции с самым типичным субстратом для SN2 – иодистым метилом. Если реакция идет и скорость ее хороша, то нужно сделать еще два теста: а ) попробовать реакцию с трет-бутилгалогенидом – не должен реагировать ни при каких обстоятельствах; б) попробовать реакцию с хиральным субстратом, например, (S)-2-иодбутаном – должны идти и давать обращенный продукт (отдельный вопрос, как это доказать, но мы вернемся к этой проблеме, когда дойдем до реакций с оксидом углерода).
Таких комплексов в химии переходных металлов довольно много. Большинство из них несут отрицательный заряд, и имеют поздний переходный металл в конфигурации d10 или d8. Вот, например, парочка особенно знаменитых, так называемых карбонилатов. Сразу видим некоторое разнообразие – металл может иметь разные степени окисления, но в пределах так называемых низковалентных форм. Видим, что число d-электронов уменьшается на два, и степень окисления тоже – замещение представляет собой окислительно-восстановительный процесс, в котором металл окисляется, а значит является восстановителем, а восстанавливается атом углерода, который меняет электроотрицательную уходящую группу на атом металла.
И видим еще одну, довольно удивительную вещь – исходные комплексы 18-электронные, а следовательно координационно-насыщенные, а продукт приобретает еще один X-лиганд. В зависимости от нашей испорченности мы можем расценить это как издевательство и признак ущербности самого понятия “координационная насыщенность”, а можем и не расценить, посчитав, что раз металл умудрился раздобыть себе еще один лиганд без изменения счета валентных электронов, то это его проблемы, а не наши. Был насыщенным и остался насыщенным – съел, и не заметил. Но ясно, что само понятие координационной насыщенности не избавляет нас от внимательного отношения к реакциям таких комплексов – оказывается, они вполне могут приобретать новые лиганды в реакциях с электрофилами (на всякий случай, если забыли – нуклеофил всегда реагирует с электрофилом, и в реакции двух кто не нуклеофил, тот электрофил, и наоборот).

Но если не в группах, то в самих рядах закономерности проявляются вполне ощутимо, только здесь в сравнение приходится брать не совсем одинаковые, а изоэлектронные частицы. Вот, например, в первом ряду металлы 8-10 групп образуют тетракарбонилы, но у металла 10 группы это нейтральная молекула, у металла 9-й это моноанион, а у металла 8-й это дианион. Дальше продлить этот ряд просто так не получится, так как для 7 группы это уже будет трианион, и вместо этого проще добавить еще один карбонил и получить частицу [Mn(CO)5]–, которая не будет прямым аналогом тетракарбонилатов. Вообще, таких анионов, так называемых карбонилатов, известно много, вплоть до ранних переходных металлов аж до 4-й группы типа [Hf(CO)6]2-, но практической ценностью в том, что касается органической химии обладают только производные поздних переходных металлов. В ряду закономерность простая – нуклеофильность падает несмотря на то, что количество d-электронов не изменяется, их доступность для нуклеофильной атаки на электрофил уменьшается по обычной причине – чем дальше по ряду (периоду), тем больше заряд ядра и компактнее электронные оболочки.

На этом пока остановимся. Примем к сведению, что некоторые комплексы низковалентных переходных металлов могут быть настоящими нуклеофилами в SN2 замещении на насыщенном атоме углерода, и такие реакции подчиняются всем закономерностям механизма SN2.
Окислительное присоединение
Многие нейтральные комплексы также умеют взаимодействовать с органическими электрофилами, но реакция протекает с одним принципиальным отличием от уже рассмотренного настоящего нуклеофильного замещения. Типический (можно даже сказать еще пафоснее – архетипический) пример такого поведения дает знаменитый комплекс Васка (или Васки – нужно ли склонять эстонскую по происхождению фамилию Vaska? – потом разберемся). Комплекс иридия (1+) реагирует с множеством соединений с разрывом σ-связей, при этом обе половинки садятся на иридий в виде лигандов. Вот, например, реакция с иодистым метилом. Самое яркое отличие от нуклеофильного замещения именно в этом – уходящая группа остается на металле, обе части субстрата становятся лигандами. Электронные счет при этом увеличивается на 2, но степень окисления металла и число d-электронов изменяются точно так же, как в нуклеофильном замещении. Реакции такого типа называют реакциями окислительного присоединения, имея в виду то, что координационно-ненасыщенный комплекс металла присоединяет новые лиганды, при этом металл окисляется, увеличивая степень окисления на 2.
Комплекс Васка и похожие комплексы других металлов реагируют с алифатическими галогенпроизводными, довольно строго предпочитая иодиды, При этом они реагируют и с третичными иодпроизводными, но очень не любят хлорпроизводные, даже метилхлорид. Уже одно это показывает, что механизм этой реакции не может быть SN2. 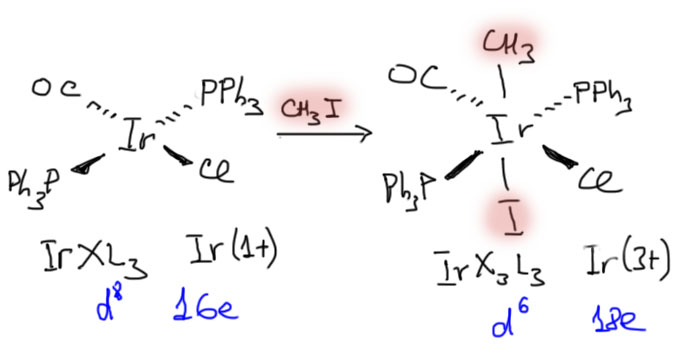
Механизм такого превращения включает перенос электрона
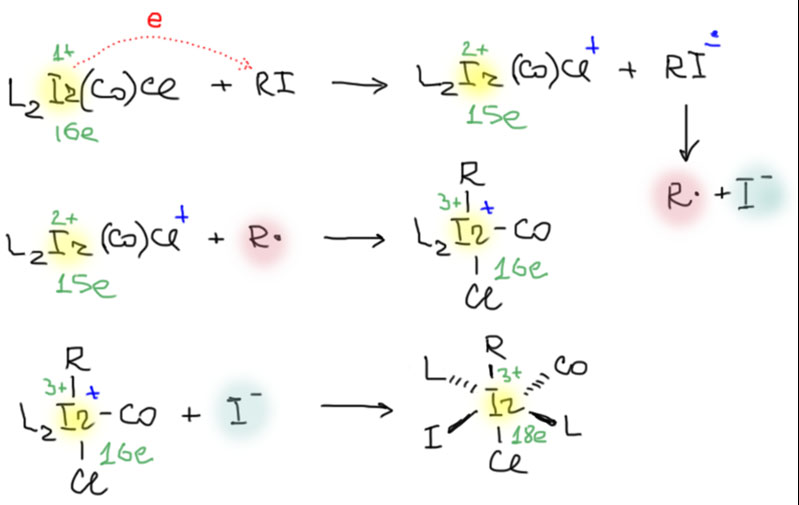
Иридий в этой реакции – двухэлектронный восстановитель. Попробуем нарисовать, что будет, если комплекс работал бы как обычный металл-восстановитель, просто передающий электроны поштучно другой молекуле (а электроны могут передаваться между молекулами только поштучно, такова природа электронов – даже если степень окисления меняется на 2, передается не пара, а один плюс один). Аккуратно напишем у каждого комплекса параметры металла (зеленым цветом – формальную степень окисления и число электронов на валентной оболочке, синим обозначим фактический заряд комплекса, там где он не ноль). Исходный комплекс 16-электронный с иридием(+1).
Шаг 1 – передаем электрон алкилиодиду, ФСО увеличивается до +2, возникает заряд +1, а число электронов становится нечетным – 15. Алкилиодид принимает электрон и превращается в анион-радикал с одним лишним электроном на связи C-I. Ослабленная лишним электроном связь немедленно рвется, и три электрона расходятся на стороны – один на углерод, два на более электроотрицательный иод. Комплекс иридия с нечетным числом электронов – фактически радикал. Обратим внимание на то, что в химии металлов не обозначают радикальный статус точкой, а просто держат это в уме – нечетное число электронов подразумевает наличие неспаренного электрона (то, что комплексы с четным числом электронов могут быть высокоспиновыми, то есть тоже радикалами, полезно не забывать, но в химии металлоорганических комплексов настоящие высокоспиновые комплексы встречаются редко из-за специфики лигандов).
Итак, образующийся при переносе одного электрона комплекс имеет и заряд и неспаренный электрон, и неполную оболочку, поэтому немедленно забирает и радикал и анион, в любом порядке (скорее всего, это вообще происходит с огромной скоростью, так три частицы – комплекс, радикал и анион моментально образуются рядом друг с другом, как иногда говорят «в клетке растворителя», мы к этому еще вернемся). Забирая радикал, увеличивает на 1 и ФСО и число электронов. Почему? Потому что присоединяет радикал с одним электроном, а формально отдает новому лиганду два в соответствии с ионной схемой дележки электронов. Итого степень окисления металла увеличивается на 2. Если у вас возникли проблемы с вторым электроном, и повышение степени окисления при присоединении радикала кажется какой-то абстрактной формальностью, проделайте то же самое, но сначала в явном виде передав один электрон от металла к радикалу, и затем присоединив получившийся анион. Будет то же самое, но окислительно-восстановительная природа процесса станет очевидной.
Результирующий процесс многостадийный, включающий стадию одноэлектронного переноса и образования радикалов. Это не очень хорошо, потому что где радикалы, там всякие побочные реакции и обычно низкая селектвность. Но, как мы скоро увидим, эта реакция может идти и по-другому.
Прямая аналогия из химии непереходных металлов
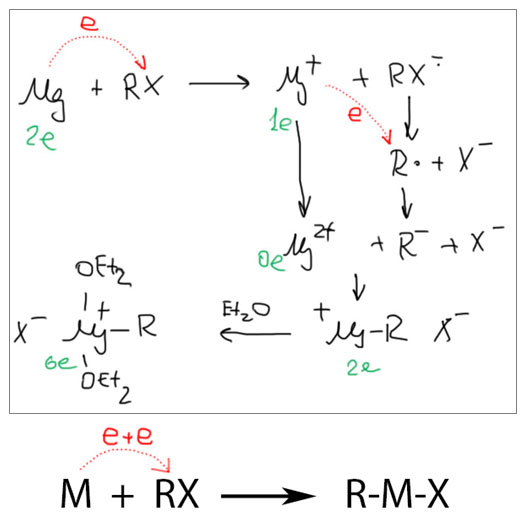
Ой, а мы это уже где-то видели! Правильно, это похоже на механизм образования реактива Гриньяра, который иногда рисуют в учебниках. Только там металл непереходный – магний, и валентная оболочка у него всего s да три p: итого октет, как и положено непереходному элементу. В исходном состоянии атома магния два s-электрона, отдаем их быстро один за другим, получаем ион магния Mg2+ с пустой оболочкой, присоединяем два лиганда R и X, получаем четыре электрона в оболочке – маловато (даже два, если учесть, что связь магний-галоген, вероятно, чисто ионная). Остаток добиваем молекулами донорного растворителя, эфира или ТГФ. Теперь понятно, почему для образования реактивов Гриньяра так важны эти растворители, без них дело не идет – непереходным металлам, как и переходным не чуждо понятие координационной ненасыщенности, и переживают они ее даже еще болезненнее. Только в мире главных подгрупп идеалом насыщения является 8 электронов (октет), а в мире переходных потребности намного выше и насытить их может только 18 электронная оболочка.
Связь углерод-галоген разрывается, когда на разрыхляющую орбиталь попадает лишний электрон
Эта аналогия неслучайна, мы имеем дело с действительно одной из самых общих реакций металлов – переходных и непереходных, ранних и поздних переходных и металлов 12 группы (группы цинка), и даже f-элементов, но последние мы договаривались не трогать. Металл или его комплекс отдает два электрона поштучно галогенпроизводному, и немедленно собирает осколки галогенпроизводного в свою координационную сферу, образуя металлорганическое соединение. Когда металл неспособен на передачу двух электронов, например, когда это щелочной металл, то расходуется два эквивалента металла на эквивалент RX, из них только один превращается в металлоорганическое соединение, а второй просто в противоион для галогенида. Посмотрим повнимательнее, почему это происходит, и в чем разница между хлор- и иодпроизводными.
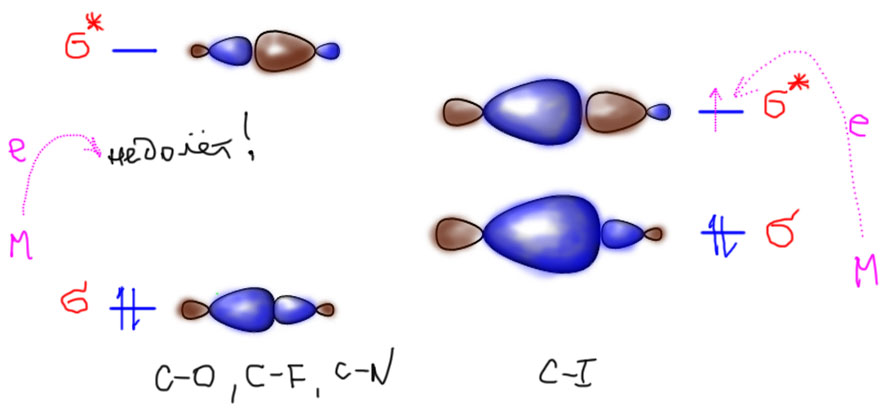
Связь углерод-галоген, как и положено σ-связям, обслуживается парой электронов на σ-связывающей орбитали. Есть и σ*-разрыхляющая орбиталь, в нормальном состоянии молекулы пустая. Как только на нее попадает электрон, связь, как и положено, разрыхляется (не зря же орбиталь так назвали), ослабевает, рвется. Лишний электрон при этом отходит более электроотрицательному атому, галогену.
Разница между связывающей и разрыхляющей орбиталями (HOMO-LUMO gap, зазор ВЗМО-НСМО) очень сильно зависит от атомов на концах этой связи. Чем связь короче, тем эта разница больше. Поэтому связи между элементами вверху групп (C-C, C-N, C-O, C-F) вообще не принимают лишние электроны – разрыхляющая орбиталь у них очень высоко. Связи между элементами внизу групп (хоть один элемент должен быть таков, как раз C-I) очень легко принимают лишние электроны – разрыхляющая орбиталь у них низко. Все. что между ними (C-Cl, C-Br) тоже рвется, но требует более сильного восстановителя. Из того, что мы уже знаем – комплекс Васка для этого слабоват, а вот металлический магний – вполне, хотя и Гриньяры гораздо легче получаются из иодпроизводных, чем из хлорпроизводных.
Как видим, этот процесс – его называют восстановительным внедрением металла по связи C-X – имеет нечто общее с нуклеофильным замещением, но таковым не является. В 1980х такой механизм (его называли SET-enabled SN – то есть нуклеофильное замещение через одноэлектронный перенос, по-русски ОЭП) пытались предложить как общий и для нуклеофильного замещения, но в конце концов победило мнение, что достаточных данных для этого нет, и привычный и классический механизм SN2 ломать не надо, и речь может идти только о двух принципиально разных механизмах. Особенно большой интерес к механизму с участием ОЭП проявляли в связи с супернуклеофильностью, что вроде бы позволяло понять, отчего так сильны нуклеофилы, производные переходных и непереходных металлов, ведь они, ежу понятно, несомненно очень хорошие восстановители. И здесь революции не случилось, и в 21 веке принято считать нуклеофилы и восстановители двумя принципиально разными типами реакционной способности.
Стереохимические последствия замещения через перенос электрона
Как доказать, что замещение идёт через перенос электрона и промежуточное образование радикалов. Это старая проблема, которая вызывала не просто бурные, а даже в прямом смысле дикие споры и раздоры в 1970-80-е годы. На первый взгдяд, проблема простая. Если радикалы образуются, то
а) их можно как-то поймать, для этого есть радикальные ловушки, соединения, которые легко реагируют с радикалами, и каким-то образом проявляют факт такой реакции;
б) реакцию можно замедлить или совсем задавить радикальными ингибиторами – соединениями, превращающими активные радикалы в малоактивные;
в) изучить стереохимические последствия реакции.
Посмотрим на вот этот последний приём, потому что он многим кажется самым красивым – ведь красива сама стереохимия. Реакции комплекса Васки и его аналогов весьма интенсивно изучали в начале 1970-х, когда казалось, что где-то около этого комплекса и лежит интересная новая химия. Надо сказать, что сам исходный комплекс Васки немного недотягивает до приемлемого уровня реакционной способности, но у же в те времена поняли, что донорность металла можно здорово повысить с помощью донорных лигандов. Уже тогда возникла идея, что нужно от триарилфосфинов переходить к фосфинам с одной или более алкильными группами. Такие группы закономерно снижают способность фосфина к back-donation (π-кислотность), и увеличивают σ-донорность. С такими аналогами комплекса Васки идет замещение с хорошей скоростью и в бромпроизводных. И тогда стало возможно изучать стереохимию замещения. Ральф Пирсон (R. C. Pearson, W. R. Muir, J.Am.Chem.Soc., 1970, 92, 5519), хорошо известный своими важными работами по теоретическому обоснованию теории ЖМКО (если вы их не читали, поделюсь субъективным мнением, что обоснования не получилось, но человек очень старался) попробовал оптически активный бромпропионат в реакции с более донорным аналогом комплекса Васки. Получил оптически активный комплекс, но вместо того, чтобы пытаться определить его конфигурацию (а это непросто – напомню, что обычный рентгеноструктурный анализ это сделать не может – он не различает энантиомеры), просто разложил его бромом. Здесь мы в очередной раз сталкиваемся с довольно зыбкими основаниями, на которых стоят многие классические исследования – исследователь пытается определить нечто в последовательности реакций, и свято при этом верит, что что-то важное происходит только в той стадии, которая его интересует. Например, в этом случае он считает что стереохимически важное событие происходит только на стадии замещения брома на металл, а дальше все определено, сам комплекс стереохимически стабилен, а реакция с бромом происходит с чистым сохранением конфигурации. Строго говоря, это если из чего-то и следует, то только из аналогий, и весьма отдалённых. Запишем то, что наблюдал Пирсон (только я припишу исходному определённую конфигурацию, в статье этого нет, а искать мне лень, хотя вполне можно найти, какой энантиомер имеет такое вращение). 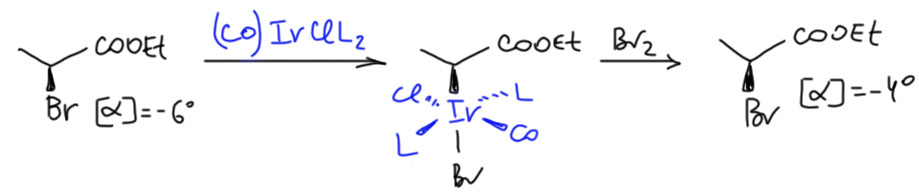
Как видим, Пирсон считал, что он наблюдал почти полное сохранение. Хотя в этом месте более современный химик может воскликнуть: “Как можно использовать такую неудачную модель с таким слабым удельным вращением!”. При таких небольших величинах вес ошибки становится неприемлемо большим. А если ошибкой пренебречь, и считать величины измеренными с хорошей точностью, то -4 по сравнению с -6, это существенно меньше и показывает значительную потерю оптической чистоты, а не сохранение. А если учесть неопределённость ошибки, то придётся принять, что степень потери оптической чистоты так и вовсе неизвестна.
Другие исследователи не стали воспроизводить данные Пирсона. Человек, далёкий от экспериментальной науки, может сильно удивиться – а почему бы не подтвердить или опровергнуть результаты, если это так важно. Но нет, так не принято – не принято делать и писать статьи, вся суть которых сводилась бы к опровержению результатов другого исследователя, как именитого (не хочется связываться), так и малоизвестного (смысла нет, никому не интересно). Другие исследователи делают другие работы. Вывод из этих работ немного странен – реакции комплексов типа Васки с галогенпроизводными с замещением на стереогенном центре приводят к неопределённому стереохимическому результату – или к частичному сохранению, или к частичному обращению, но всегда с частичной потерей конфигурации. Это довольно типичный результат для реакций с участием частиц типа радикалов или катионов. Сами по себе эти частицы, если образовались, не обладают хиральностью – если атом, на котором образовалась точка или плюс до этого был стереогенным с определённой конфигурацией, в промежуточной частице конфигурация теряется. Но при образовании продукта за счет связывания радикала с радикалом или катиона с нуклеофилом оптическая активность может восстановиться или измениться на противоположную с частичной потерей, если время жизни радикала или катиона столь мало, что это связывание происходит с учетом положения частиц, возникших при образовании радикала или катиона – они не успевают изменить своего положения в пространстве, ведь движение молекул в жидкости не моментально, а происходит с конечной скоростью, которую можно грубо оценить по параметрам диффузии в данной среде.
В качестве небольшой задачи попробуйте разобраться в результатах ещё одной работы по выяснению стереохимии реакции галогенпроизводного с комплексом типа Васки.
Вопрос: сохранение или обращение
Другой крупный металлорганик Джон Осборн (ученик одного нобелевского лауреата Дж.Уилкинсона и научный руководитель другого, Р.Шрока, причём этот последний даже сварил одно из исходных для этого исследования, но в авторы статьи попасть чести не удостоился, так что, если с вами самими приключится такая грустная история – в самом начале своей деятельности сварите какое-то соединение, его используют в статье старших коллег, но вас в авторы не возьмут, не расстраивайтесь, потому что знайте, что именно так и начинал один из крупнейших металлооргаников современности, лауреат нобелевской премии 2005 года Ричард Шрок) решил выбрать более удобную модель для выяснения того, что происходит при реакции иридиевого комплекса с галогенпроизводным – диастереомер, позволяющий определять относительную конфигурацию с помощью ЯМР (J. A. Labinger, A. V. Kramer, J. A. Osborn, JACS, 1973, 95, 7908 doi: 10.1021/ja00804a080). При наличии таких объёмистых заместителей каждый диастереомер имеет единственную устойчивую конформацию, и такие конформеры отлично видно в ЯМР по вицинальным константам спин-спинового взаимодействия. Напомню вам знаменитое правило Карплуса (еще одного нобелевского лауреата, Мартина Карплуса, или, как у нас его называют по традиции, Карплюса) – вицинальные константы (имеются в виду любые, протон-протон, протон-фтор, фтор-углерод и т.п.) имеют максимальную величину, если ядра находяться в анти-конфигурации, и намного меньшую – если в гош-конфигурации, а если позабыли эти термины конформационного анализа, можете освежить на моем сайте про органику).
Итак, Осборн взял чистый эритро-диастереомер эфира достаточно богатой заместителями α-бромкислоты и провел реакцию с ещё более донорным аналогом комплекса Васки, получив смесь двух диастереомеров эфира с иридиевым заместителем (на схеме Ir означает весь комплекс с еще одним лигандом Br) в соотношении 4.5 к 1 (на схеме больший изомер наверху с соответствующими константами, меньший внизу). 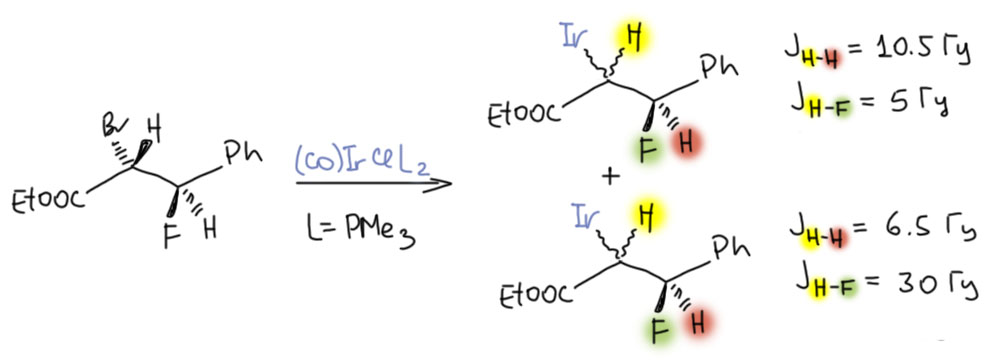
Попробуйте вспомнить конформации и разобраться в продуктах, ответив на вопрос – в этом эксперименте преобладает обращение или сохранение конфигурации? Если полезете в оригинальную статью, учтите, что там всё очень коротко и додумывать всё равно придётся.
(за полный ответ со схемами и объяснением можно получить до 20 плюсиков)
Согласованное окислительное присоединение
Итак, некоторые комплексы переходных металлов проявляют способность расщеплять связи углерод-галоген и некоторые другие через перенос электрона. Важным признаком здесь является относительная реакционная способность алкилгалогенидов – эти реакции практически не видят разницы между первичными, вторичными и третичными алкилгалогенидами, и при этом очень любят иодпроизводные, и очень не любят хлорпроизводные. Это очень характерно для одноэлектронного восстановления, для которого стерика субстрата практически не важна, но очень непохоже на классический SN2. При этом в таких реакциях не реагируют ароматические и непредельные галогенпроизводные.
Но история на этом не кончается, а только начинается. Более интересной и богатой оказалась химия d10-металлов, вначале нульвалентных представителей 10 группы – Ni, Pd, Pt. У этих металлов известно множество комплексов с таким валентным состоянием. Самый простой и удобный – тетракис(трифенилфосфино)палладий(0) охотно реагирует с разнообразными галогенпроизводными, причем особенно легко – с ароматическими иод и бромпроизводными. Подробные исследования этой реакции показали, что перед тем как реагировать с галогенпроизводным, в растворе комплекс самопроизвольно диссоциирует. В этом нет ничего странного – это просто является следствием не очень большой константы устойчивости комплексов PdL4 и PdL3 – и не означает, что диссоциация происходит полностью или даже на существенную глубину – достаточно небольшой концентрации реакционноспособного комплекса PdL2, чтобы реакция пошла, а дальше будет работать принцип ЛеШателье, так как только последний комплекс реагирует, а следовательно выходит из равновесия. Обратим внимание на то, что при диссоциации нейтрального лиганда степень окисления не изменяется, а число электронов уменьшается – образуется координационно-ненасыщенный комплекс, металл в котором жаждет вернуть утраченное. Где мои 18 электронов? 18 не обещаем, не нужно было фосфин терять, но 16 можно вполне найти, присоединив иодбензол или что-то подобное. Образуется новый комплекс, очень похожий на то, что происходит при восстановительном внедрении у того же комплекса Васка, да и реактива Гриньяра тоже. Но – никаких признаков переноса электрона и чего-либо свободнорадикального здесь нет. Да и выбор субстрата необычен – иод и бромпроизводные бензола не так-то просто восстановить – связи C(sp2)-X намного прочнее связей при насыщенном атоме углерода, хотя бы потому что они существенно короче.
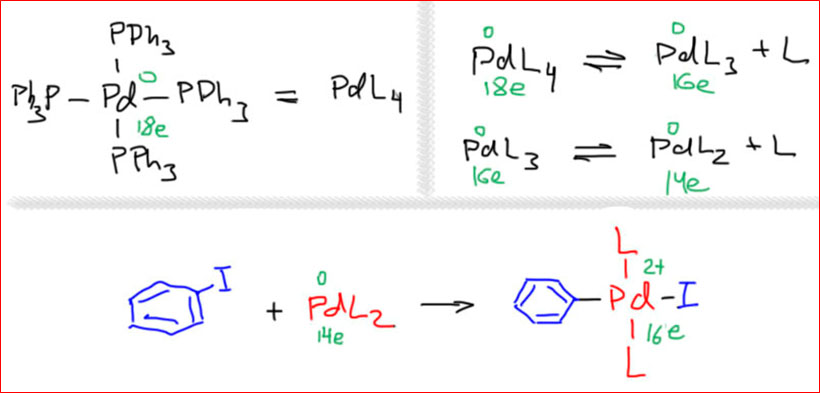
Согласованный механизм окислительного присоединения
Эта важнейшая реакция, скорее всего, является так называемым согласованным процессом. В химии переходных металлов согласованные процессы встречаются очень часто, и это одна из особенностей и сильных сторон этой химии. Напомню, что с согласованными механизмами мы хорошо знакомы – в обычной органической химии согласованными процессами являются, например, замещение SN2 и циклоприсоединение по Дильсу-Альдеру: согласованные механизмы всегда имеют одну стадию, в которой все и происходит – все, что должно порваться, рвется, и все, что должно образоваться, образуется. Переходное состояние согласованных процессов – это такой моментальный снимок происшествия, стоп-кадр: рука убийцы с ножом движется к несчастной жертве, все еще живы, и все еще можно повернуть назад. Но нет, – то, что должно случиться, случится, и труп будет лежать в луже крови.
Так образуются продукты в согласованных реакциях. Никогда не бывает ничего посредине – никаких промежуточных молекул, интермедиатов. Но перед тем, как произойдет собственно превращение, молекулы подходят друг к другу, собираются в предреакционный комплекс, ориентируются так, чтобы задействованные в превращении орбитали максимально точно расположились в пространстве, обеспечивая максимально возможное взаимодействие (перекрывание). Поэтому согласованные реакции всегда чрезвычайно точны стереохимически (все помнят , например, яркие стереохимические особенности SN2-замещения и реакции Дильса-Альдера).
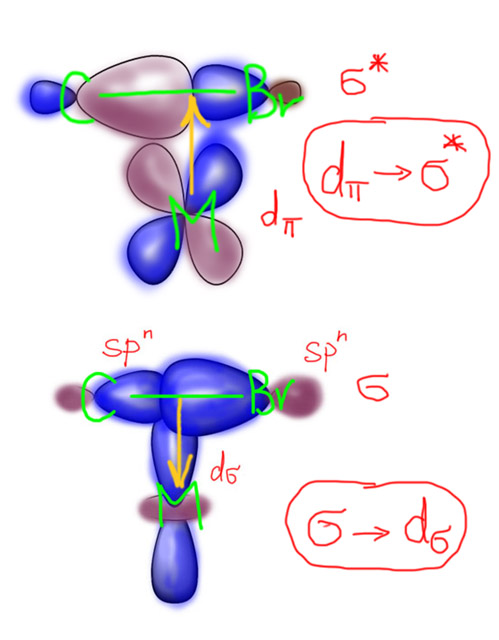
Итак, сначала происходит вхождение галогенпроизводного в координационную сферу d10-металла за счет η2-связывания галогенпроизводного через орбитали связи C-X. Фокус здесь в том, что металл с меньшим числом электронов предпочел бы связывать ароматический субстрат через p-связи ароматического кольца, образуя η2-, η4– или η6-ареновые комплексы, но богатый электронами металл выбирает для связывания акцепторный фрагмент – σ-связь углерод-галоген, обладающую, как мы это уже выяснили, очень доступной по энергии разрыхляющей орбиталью.
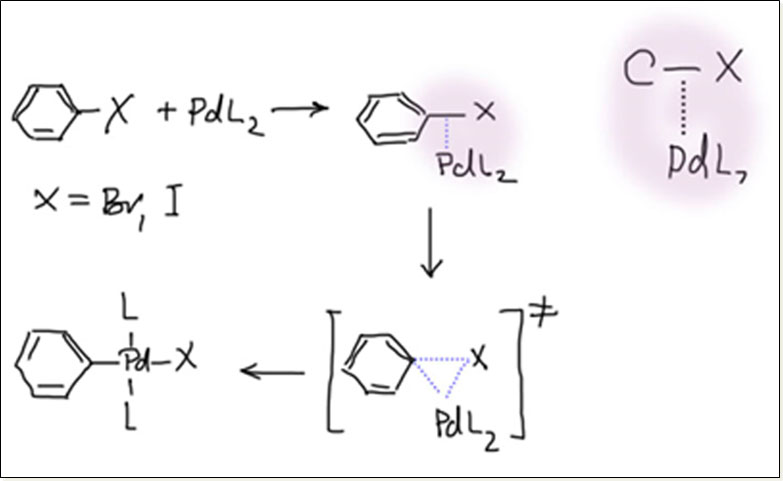
На эту орбиталь очень удобно задвинуть электроны с заполненной dπ-орбитали через механизм π-донорного взаимодействия (back donation). Обратим внимание на то, что это взаимодействие в комплексах, как правило, делает более прочной связь металла с лигандом, обеспечивая дополнительное связывание помимо прямой координационной связи. Как правило, и мы еще много раз увидим примеры этого, прямая координационная связь лиганд-металл более важна, а back-donation только добавляет небольшой или более значительный вклад в общую энергию связи металла и лиганда. Но в рассматриваемом случае относительный вклад этих взаимодействий и их роль существенно иные.
Прямая координационная связь здесь очень слаба, так как из-за отсутствия вакантных d-орбиталей должна обязательно использовать более высоколежащие s или p-орбитали валентной оболочки металла, или, точнее, гибридные орбитали с участием s или p (на рисунке показана условная доля какой-то орбитали, подходящей для σ-связывания). А вот обратное донорное взаимодействие от металла на лиганд работает на полную катушку, но при этом разрушает старый лиганд, создавая два новых. Это странный, на первый взгляд, случай, когда π-донорное взаимодействие преобладает. Вполне очевидно, что при этом C-X связь разрыхляется – на ней оказывается лишняя электронная плотность, а мы знаем, что связи C-галоген немедленно рвутся уже от одного лишнего электрона. Собственно, это то же самое, что происходит в только что рассмотренном механизме окислительного присоединения через перенос электрона (или, что то же самое, восстановительного внедрения металла по связи углерод-галоген) с одним принципиальным отличием – там металл и галогенпроизводное перебрасывались электронами на расстоянии, а здесь все это происходит в координационной сфере металла, и в завершении процесса обе части бывшего галогенпроизводного оказываются σ-связанными X-лигандами в новом комплексе.
Невероятная мощь никеля(0) в окислительном присоединении
Когда мы обсуждаем окислительное присоединение и основанные на нем процессы, то в основном говорим о палладии, а никель все время маячит за спиной как такой бедный родственник. Но это не совсем так. Никель – очень интересный металл, и работ про катализ комплексами никеля становится все больше. Одной из замечательных особенностей никеля является его выдающаяся реакционная способность в окислительном присоединении. Палладий, как мы знаем, отлично берет связи C-I, C-Br и C-O, но только в трифлатах. Связь C-Cl подчиняется палладию с помощью новых донорных фосфинов, они же помогают палладию справиться со связью C-O в менее реакционноспособных тозилатах. Но где-то здесь и лежит предел возможностей палладия. В то же время нульвалентный никель не только легко справляется со связью C-Cl, но и способен окислительно присоединять чрезвычайно инертную связь C-F. В последние 10 лет появилось немало работ, в которых разработаны протоколы на основе Ni(0) и донорных лигандов для кросс-сочетания фторпроизводных. Более того, Ni(0) может присоединять и связь C-O в совсем уже нереакционноспособных метокси-производных. Примеры таких реакций приведем при обсуждении реакций кросс-сочетания