Краткие разборы заданий 2-й части квеста
По большинству заданий второй части квеста 2023 года написаны подробные разборы, и материала оказалось так много, что я решил его просто так не выкладывать, потому что в этом просто так не разобраться, имея только номер задания. Через некоторое время я начну делать указатель к таким страницам, чтобы можно было выбирать материал по реакции, лиганду, протоколу и другим конкретным особенностям. А пока вот краткие разборы ко всем заданиям этого года.
G-1
Здесь тандем (авто-тандем) двух реакций Стилле, межмолекулярной и внутримолекулярной. Используется сокатализ комплексом Либескинда CuTC, и это очень важное дополнение к реакции Стилле, а в чём его суть и какую роль здесь играют добавки разберём в полном разборе. Анциллярный лиганд трифенилфосфин. Многие не замечают трифенилфосфин, как будто это что-то совсем бесполезное, типа, подумаешь, старьё какое, просто другого предкатализатора не было, вот и взяли этот. Но какие у нас права обижать этот исторический лиганд с такой историей, что все патентованные и непатентованные новинки умрут от зависти. Лиганд, который в одиночку создал и кросс-сочетание, и немало других важнейших процессов. Присутствие трифенилфлсфина в координационной сфере точно влияет на скорости кататлитических реакций, а то, что иногда можно и без него, никак не отменяет тот факт, что большинство предпочитает не рисковать, и трифенилфосфином не брезгует. В тандеме TON это грубо средняя оценка приблизительно 2 (один межмолекулярный, один внутримолекулярный). TOF тоже грубая оценка – меньше 1 цикла в час. Реакция диастереоспецифическая – сохранение конфигурации (цис по одной двойной связи, транс по другой).
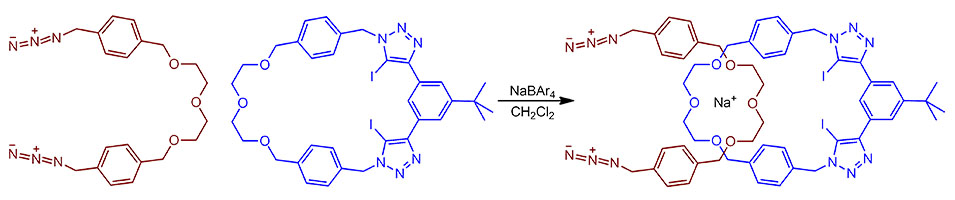
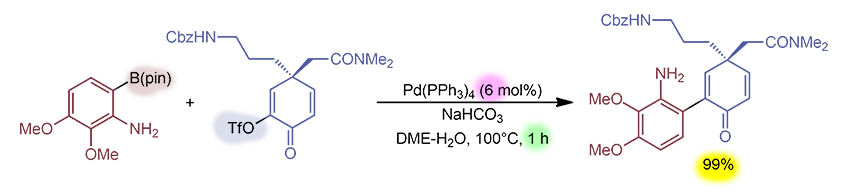
G-2
Очень интересный пример применения клик-реакции CuAAC для сборки сначала макроцикла и затем катенана, с использованием комбинированного темплатного эффекта. При выходе катенана в 20% (это данные из доп. информации, в статье данные другие и по выходу и по времени) и загрузке катализатора в 50 моль% на две сшивки, реакция фактически некаталитическая, поэтому TON/TOF считать не будем. В реакции используется анциллярный лиганд TBTA, предложенный Шарплессом и Фокиным в 2004 для стабилизации Cu(1+). Саму реакцию выполняют в очень интересном режиме. Сначала одну часть будущего катенана запихивают во вторую, используя равновесие образования комплекса с катионом натрия (противоион специально выбран большой и очень слабо взаимодействующий с катионом, поэтому несчастный катион особо усердно ищет взаимодействий, и находит их в двух фрагментах крауна -так и работает темплатный эффект. Поскольку мы не совсем наивные люди и кое-что в химии понимаем, нас не должна удивлять такая картинка, как будто тут все так идеально друг в друга входит. Понятно, что в реальности там довольно широкое распределение и конформаций и взаимного взаимодействия фрагментов, и что очень слабым взаимодействием катиона натрия с кислородами (это чистая электростатика, здесь нет никакой ковалентности) не под силу так аккуратно все собрать, но тем не менее, они могут обеспечить существенное превышение вероятности вхождения новой цепи в уже готовый макроцикл, а авторам работы ровно это и надо.
Выдержав некоторое время для установления равновесия, в систему добавляют остальные компоненты клик-реакции. И здесь метафора клик совсем удачна – тем цепочкам, которые, может юыть сдуру влезли в макроцикл, думая, что сейчас там всё обнюхают и выдезут, быстренько – клик! – и навесили триазольный замок сначала с одной стороны – с ним уже не вылезешь, а затем уже без большой спешки замкнули второй макроцикл. Вуаля – катенан! И не случайно образовавшийся просто по теории вероятностей (когда рулит энтропия), а так как будто хотели катенан – и получили катенан, побороли энтропию энергие комплексообразования. Ну, реальность малость прозаичнее, никто никого не победил, но вероятность повысить удалось хорошо – энтропию все же слегка попросили уступить хоть немного.
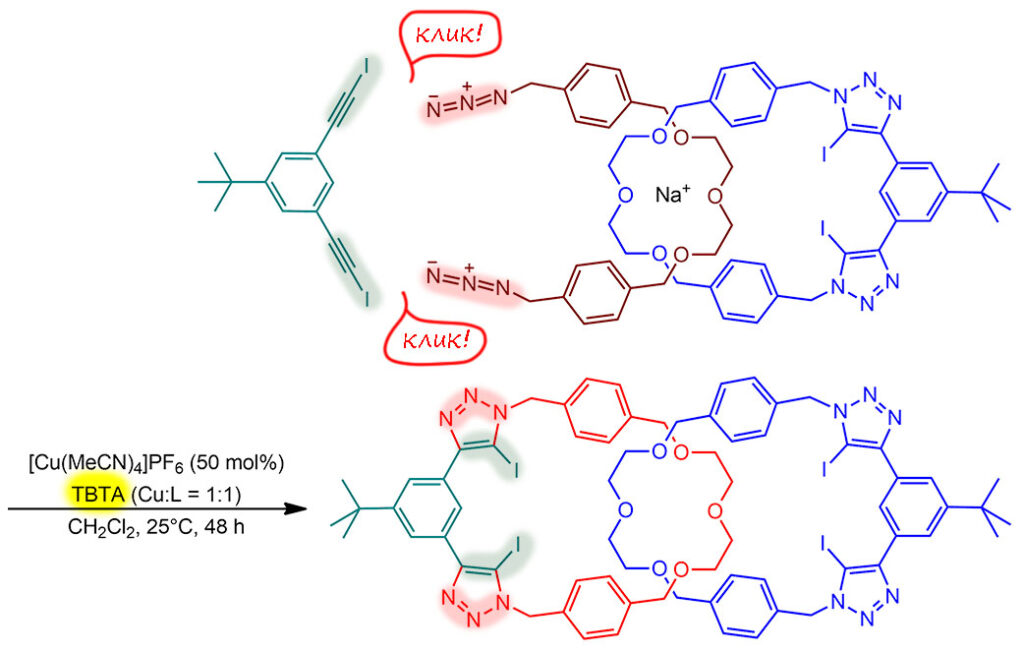
И в очередной раз обращаю внимание, что то, что кажется двумя одинаковыми реакциями, на самом деле очень разные реакции. Первый клик просто вешает ацетиленовый фрагмент на конец цепи, а второй это макроциклизация. Поэтому если бы мы могли наблюдать за ходом процесса, мы бы наверняка увидели, чо первая реакция идет быстрее и с большим выходом, а весь процесс лимитирует вторая реакция.
G-3
Это реакция KTK, катализируемая комплексом палладия. Анциллярный лиганд – трис-третбутилфосфин, стандартный донорный объёмистый лиганд, здесь он наверняка играет активную роль, потому что авторы статьи особо пишут о том, что именно он дал лучший результат. Предкатализатор Pd(tBu3P)2. TON 10-11, TOF меньше 1 цикла в час. Реакция кросс-сочетания ненаправленная, хотя магнийорганика для нее получена направленной реакцией карбомагнезирования. В подробном разборе обсудим этот способ получения магнийорганики для КТК. Реакция диастереоспецифическая, потому что идет с сохранением конфигурации, и это следствие механизма реакции. Для диастереоспецифической реакции параметры селективности указывать не надо. То, что в описанном эксперименте получается смесь диастереомеров E:Z 88:12, могло бы говорить о диастереоселективности (плохой!), но мы уверены, что это следствие потери конфигурации субстратом, хотя, возможно, это связано с неудачными условиями реакции кросс-сочетания. Оба мнения имеют право на жизнь.
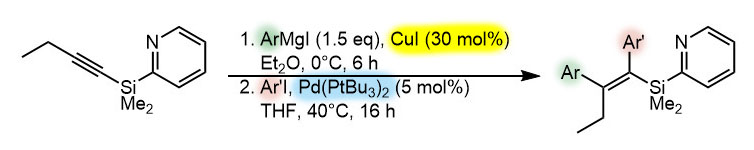
G-4
Кросс-сочетание, реакция Судзуки-Мияуры во вполне классическм исполнении, только взята не бороновая кислота, а ее пинаконовый эфир, появившийся прямо из борилирования. Как видим, это никак не сказывается ни на выборе протокола, он вполне обычный, даже слишком, ни на выходе – выход количественный, даже немного неприличный, но раз получили, значит получили. Это тем более важно, так как субстраты кажутся довольно проблемными. Со стороны бороната свободная амино-группа в орто-положении. Со стороны трифлата енон, правда неенолизуемый, и с защищенными функциями. Но такой субстрат все равно должен быть чувствителен к основности, поэтому, скорее всего, взяли очень щадящее основание, гидрокарбонат, причем водный, и все получилось блестяще. TON, как обычно в синтезе, невелик из-за солидной загрузки предкатализатора, приблизительно 15 циклов, и все это за час, то есть TOF 15 циклов в час уже для нас довольно приличный, реакция достаточно быстрая, хотя идет при нагревании. Больше тут нет ничего, ни направленности, ни стереоселективности, ни осложнений.
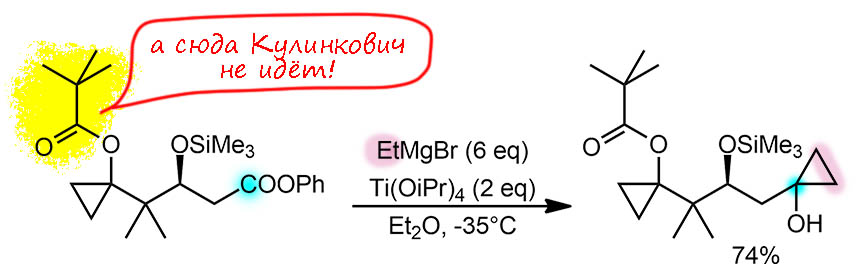
G-5
Вполне типичная реакция Кулинковича из синтетической работы самого Кулинковича. Просто формируется циклопропанол на месте сложноэфирной группы в соединении, где уже есть одно циклопропановое кольцо, скорее всего раньше тоже сформированное Кулинковичем. Интерес в этой реакции еще и в том, что в исходном субстрате две сложноэфирные группы. но реагирует только одна. Триметилацетильная (пивалоильная) группа в реакции не участвует, что делает такую группу хорошей защитой в реакции Кулинковича. Инертность, скорее всего, обязана стерике; титан в этом смысле вполне типичный переходный металл, сильно боящийся стерики. Реакция некаталитическая, поэтому считать ничего не будем. Всё остальное тоже нерелевантно.
G-6
Две последовательные реакции или палладий-катализируемое CH-арилирование или внутримолекулярный Хек. Эти реакции непросто различить, поэтому подробнее обсудим в полном разборе. Реакции кажутся одинаковыми, но первая реакция идет в субстрате с повернутыми друг относительно друга частями, а второй- когда они уже стали плоскими и прямо наезжают друг на друга. Поскольку активировать надо C-Cl, используется сильнодонорный анциллярный лиганд трициклогексилфосфин. Загрузка предкатализатора случайно или неслучайно точно соответствует выходу, поэтому реакция некаталитическая, стехиометрическая по палладию, то есть TON равен единице (напомню, что TON считается на атом палладия, и хотя здесь две реакции, каждая из которых представляет собой цикл, но каждый атом палладия участвует только в одном. Забавный парадокс, не правда ли, – цикла явно два, а TON – единица. Хороший повод запомнить, что TON – величина удельная и чисто оценочная. TOF соответственно – цикл за 2.75 часа, или треть цикла в час. А треть цикла это что такое? Не будем о грустном, это оценочные величины для грубого сравнения процессов, а не для размышлений. Стереохимии тут нет. Направленность есть – она определяется структурой исходного и пространственной сближенностью конкретных центров.
G-7
Очень большая молекула собирается за один прием шестью реакциями Соногасиры. В самой работе, просто удивительно неряшливо написанной, с тяжёлой путаницей и несоответствиями между текстом статьи и доп.информации, утверждают, что это реакция Кадьё-Ходкевича, но это не так, та реакция идет в присутствии только Cu(I), а здесь почти типичные условия Соногасиры, в котором вполне можно использовать и галоалкины. Почему почти – потому что в классическом Соногасире редко используют бесфосфиновый вариант, да еще и с dba-комплексом палладия, да еще при комнатной температуре – в таких условиях вспомогательный dba-лиганд скорее все портит, потому что сильно не торопится освобождать координационные места. Другой странностью является очень малая загрузка, которая дает TON более чем 150 циклов (с учетом числа реакций на молекулу), TOF приблизительно 4 цикла в час. Смотрим на молекулу и видим очень большие стерические проблемы при ее сборке, даже при учете того, что тройные связи позволяют свободное вращение и каждый из фрагментов может быть развернут боком, но это говорит только о том, что такую молекулу собрать можно, но точно не без больших сложностей, особенно на последних стадиях. Еще одна странность – точное соотношение исходных 6:1 – так практически никогда не делают, когда нужно достичь полноты сочетания – всегда берут хотя бы небольшой избыток второго реагента. Здесь конечно выход не 100%, но он довольно высокий для такой реакции, и нет никаких указаний на то, что в реакции образуются неполные продукты сочетания, которые обычно очень трудно отделить от полных. Химия так устроена, что мы не можем только на основании наших сомнений отвергать результат, но сомневаться нам никто запретить не может – я бы не посоветовал это пытаться воспроизвести, только время потеряете. Дальше там кстати все еще страннее.
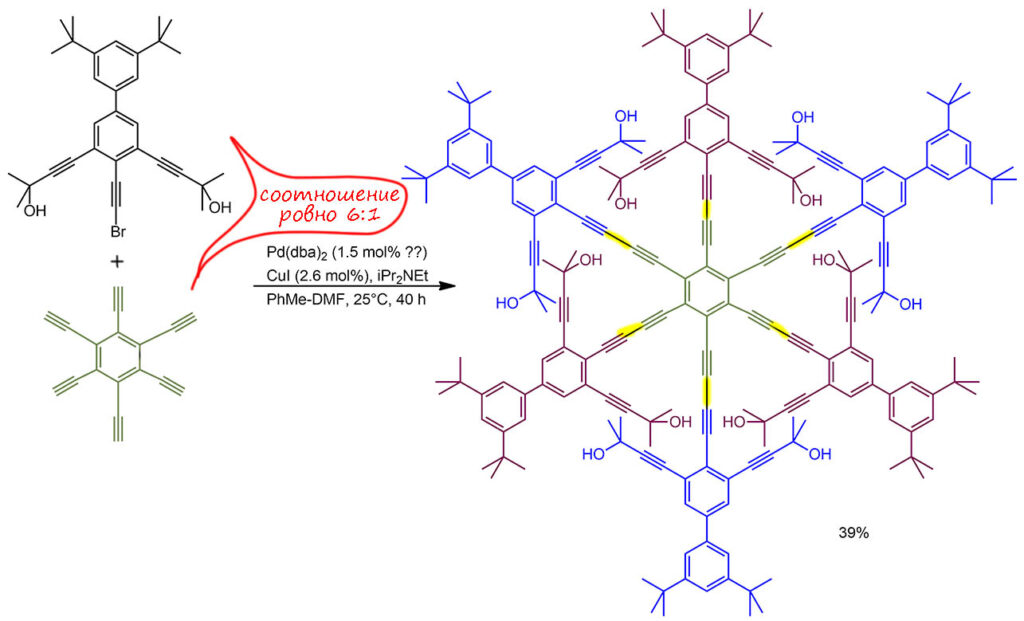
G-8
Очень интересный пример применения катализируемого комплексами золота нуклеометаллирования для синтеза очень модных нынче циклических пептидов с неаминокислотными вставками. Сама реакция предельно проста и стандартна – присоединение амина к тройной связи с экзо-диг-циклизацией, и самопроизвольной перегруппировкой енамина в имин. В статье продукт сразу же гидрируют боргидридом натрия в просто амин. Предкатализатор – один из самых популярных, катионный комплекс золота с бакуолдовым анциллярным фосфином JohnPhos, но берут его даже не в стехиометрических, а в избыточном количестве – здесь важнее надежность, чем экономия на золоте, потому что и так понятно, что сами такие соединения сильно дороже золота; можно было бы даже сказать, что комплекс золота здесь – самое дешевое, если бы не добавка ещё более дешёвой четвертичной аммонийной соли, стреляйте не пойму зачем, а объяснение в статье малость наивное и явно пренебрегающее тем очевидным обстоятельством, что комплекс золота в присутствии бромид-иона перестанет быть катионным. Вполне возможно, что такое смягчение электрофильности золота просто приводит к большей избирательности, ведь сколько бы мы ни обзывали золото карбофильной кислотой Льюиса, но оно не совсем пренебрегает другими нуклеофилами, а в таких больших молекулах нуклеофильных мест очень много. Катионное золото, видимо, цепляется за другие и отвлекается от своей основной, карбофильной работы. А нейтральное, более координационно насыщенное, действует с большей разборчивостью и почем зря за чужими неподеленными парами не клеится. Поскольку реакция очевидно стехиометрическая, считать тут нечего. Стереохимии тут тоже никакой нет, потому что концевой амин специально берут из самой простой аминокислоты, глицина, и нет даже случайных потерь или переносов хиральности. Обратим еще внимание, что в такой макроциклизации, а размер цикла огромен (сами посчитайте), очень приличный и даже просто высокий выход. Это очередное свидетельство того, что ограничения множества конформеров увеличивают вероятность встерчи концов, а у пептидов конформационное пространство сильно сужено из-за хорошо всем известных взаимодействий, определяющих вторичную структуру белков – цепь сама сворачивается в спирали и широкие кольца, обеспечивая очень приличную вероятность встречи реакционных центров.
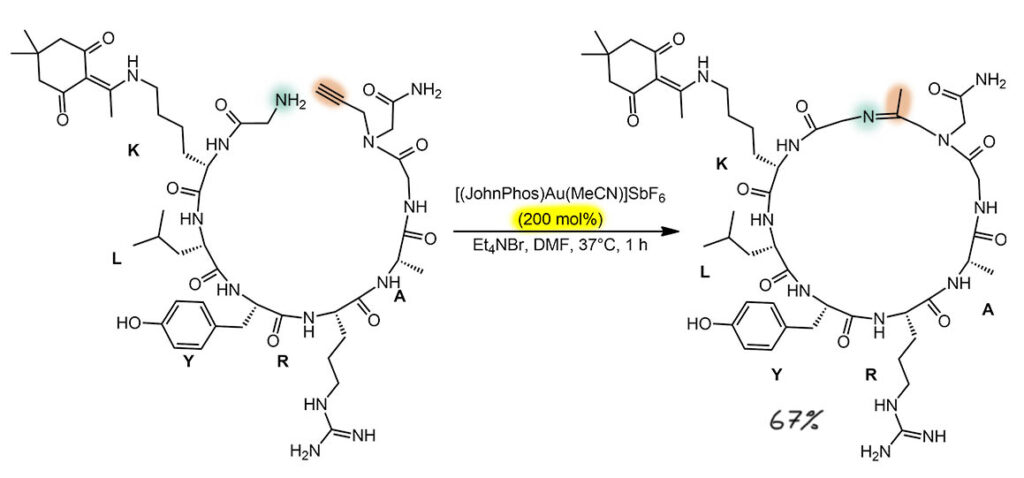
G-9
Реакция Посона-Кханда, внутримолекулярная. В ходе реакции образуется циклопентенон, приконденсированный к 7-членному циклу, и такое сочетание циклов не помогает селективности – в реакции образуется второй изомер. Поскольку в реакции Посона-Кханда первой образуется связь, расположенная напротив будущего карбонила, именно по образованию этой связи можно различить экзо и эндо-типы циклизации, и тогда мы видим, что оновной продукт соответствует экзо-диг, а минорный – эндо-диг способом. Реакция стехиометрическая с 50%-ным избытком карбонила кобальта. Не забываем, что в этой реакции нужны оба атома кобальта, поэтому мы не приводим загрузки к одному атому, как, например, для палладиевых димерных предкатализаторов. Поскольку реакцию ведут без атмосферы CO, лишний карбонил может быть источником концентрации оксида углерода в реакционной смеси. Реакция начинается с образования комплекса карбонила по тройной связи (1 час при 40°), дальше при 60° кластер превращается в продукт в виде смеси диастереомеров. В этом месте нужно посмотреть на ситуацию аккуратно, потмоу что в статье все написано мутно и скороговоркой, а методики в доп.материале еще и на непонятном языке, отдаленно напоминающем английский, но им не всегда являющимся. Поэтому может создаться впечатление, что еракция Посона-Кханда как-то затрагивает стереоконфигурацию аллильного центра. Мы об этом ничего не знаем, поэтому сильно напрягается. Не стоит оно того, всё намного проще. Конфигурация этого стереоцентра в ходе реакции не изменяется, но два диастереомера явно реагируют с разными скоростями (или выходами – мы не можем различить из представленных данных, что верно), причем ненужный, минорный диастереомер реагирует быстрее, из за чего исходное соотношение 10:1 изменяется к 5:1. Авторам синтеза нужен именно основной диастереомер, потому что это тот тип синтеза оптически активного природного вещества, в котором хиральность создается однажды в самом начале длинной цепи и далее сохраняется благодаря диастереоселективности всех последующих стадий. Или, если она плохая на какой-то стадии, приходится разделять диастереомеры и дальше отправлять только нужный. Вот это ровно случай этой стадии, и мы видим, что выход по тому соединению которое реально будет использовано дальше, всего 45%. Но остроумные авторы работы, чтобы добро не пропадало, взяли именно ненужный диастереомер, закристаллизовали его и сделали рентген, чтобы подтвердить стереохимию этого ключевого в данном синтезе полупродукта, и не тормозить основную работу.
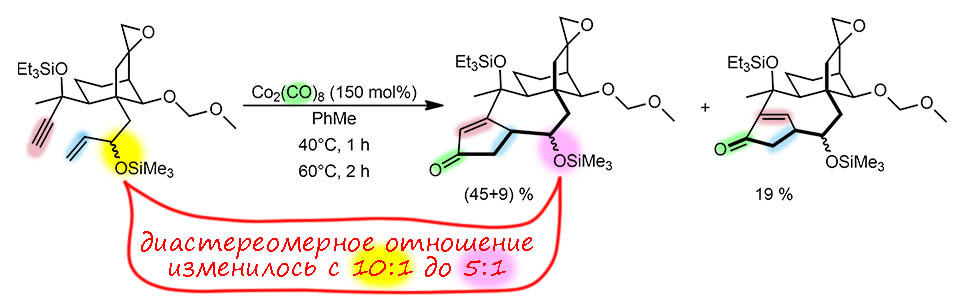
G-10
Реакция Судзуки-Мияуры в очень важном варианте – сочетание с алкильной борорганикой, которая обычно получается в том же ведре и используется без выделения. Здесь это еще и макроциклизация, образуется 24-членный лактонный цикл (макролид). Дополнительная сложность – второй участник кросс-сочетания – геминально замещенный алкенилиодид, и мы по реакции Стилле знаем, что это доставляет немало проблем. В реакции Судзуки-Мияуры проблемы немного другие, чем в Стилле, но здесь точно нужно давить гидридное элиминирование, хотя нужно понимать, что мы в таких работах просто не видим, есть побочные реакции или нет, потому что делают такие синтетические стадии (а это почти последняя в длинной цепи) с мизерными загрузками (здесь – 5 мг), и продукт в количестве нескольких миллиграмм выделяют препаративной HPLC с детектором, который фиксирует молярные массы, просто не обращая внимания на все остальное. Здесь используется типичный анцилляр для кросс-сочетаний с алкилными субстратами – dppf, но в смесь добавляют еще трифениларсин, зачем – обсудим в подробном разборе, но это стандартный протокол для таких кросс-сочетаний. Загрузка предкатализатора лошадиная, что делает всю реакцию некаталитической, и ничего считать не будем. Такая загрузка тоже определяется тем, что реакция делается на очень малое количество, и тогда просто дозировать предкатализатор сложно, проще добавить побольше, никто не осудит. Циклизация диастереоспецифическая с сохранением конфигурации двойной связи.
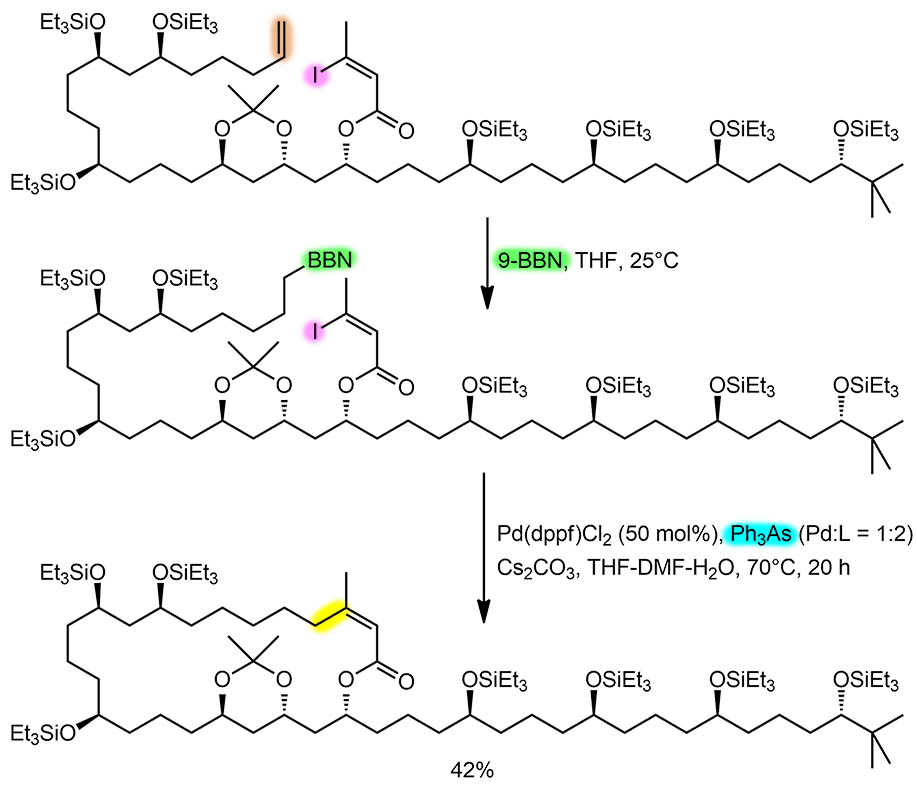
G-11
Асимметрическое циклопропанирование. В статье для этого примера даны два катализатора, один родиевый, типа “гребное колесо”, и второй, рутениевый. Но в условиях квеста дано, что в случаях, когда есть несколько примеров, нужно выбрать лучший. Лучше здесь проявил себя рутениевый комплекс, причем это рутенацикл, сделанный из хирального лиганда хорошо нам знакомой оксазолиновой серии, в данном случае это не PyOx, а PheOx, так как рутений связывает обычное фенильное кольцо, образуя металлацикл. Показанный комплекс это не катализатор, апредкатализатор, так как металл забит под завязку ацетонитрилами, часть из которых в растворе очевидно диссоциирует, освобождая место карбеновому лиганду – весь комплекс становится хиральным карбеноидом. Статья в одном из новых журналов, к сожалению, написана неаккуратно, и данные противоречивы: пришлось взять выход из эксперимента, а ee из таблицы в обсуждении, где немного другие условия и другой выход. Само циклопропанирование здесь внутримолекулярное, карбеноид генерируют из диазокетона, и он внутримолекулярно атакует двойную связь, создавая два стереогенных центра. Энантиоселективность не очень высокая, но в других примерах статьи часто и того ниже, причем рутениевые катализаторы, существенно более простые с виду, чем родиевые типа “гребное колесо”, дают более высокие энантиомерные избытки. Зато TON в реакциях циклопропанирования карбеноидами достаточно высок, 70 циклов. Да и TOF тоже ничего – 70 циклов в час, каждую минуту по циклу с хвостиком. Это заслуживает комментария – у быстрых реакций невысокие барьеры активации. Но для того, чтобы осуществилась энантиодискриминация двух подходов к прохиральному реакционному центру, должна быть заметная разница в барьерах активации двух путей. Так как барьер невысок, добиться разницы сложно, и поэтому реакцию ведут при пониженной температуре, чтобы хотя бы небольшая разница дала различие в скоростях реакций.
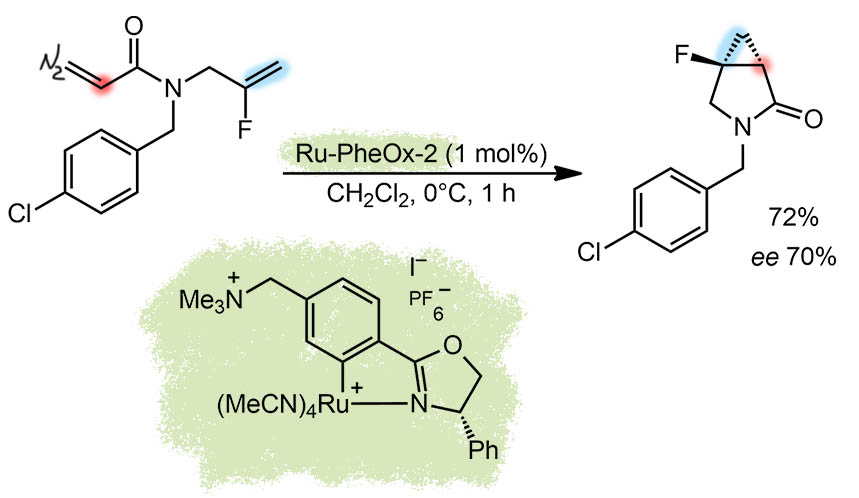
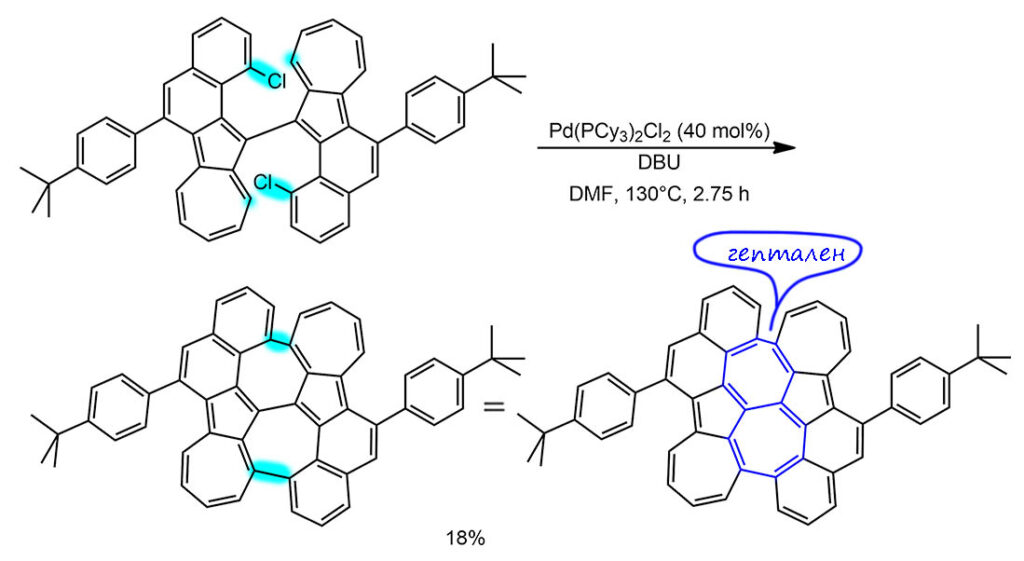
G-12
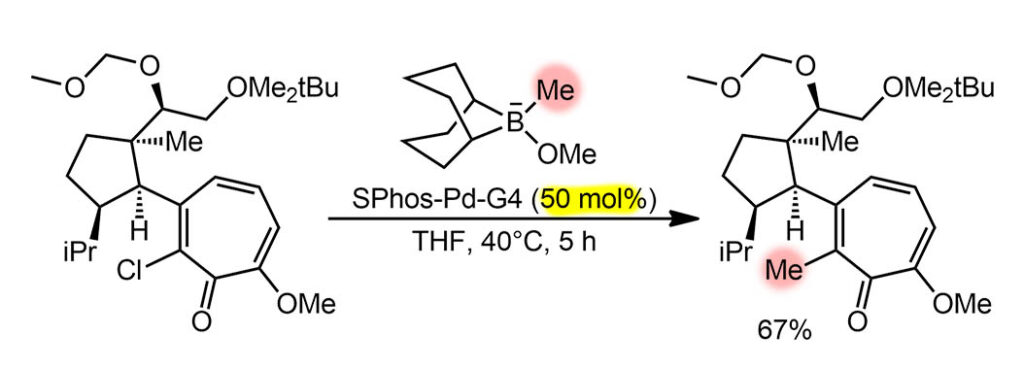
Это просто реакция Судзуки-Мияуры, но в чрезвычайно непростой ситуации. Вот, казалось бы, что может быть проще, чем воткнуть в молекулу метильную группу. Но пришлось очень сильно постараться, использовав новейшие достижения кросс-сочетания и тонкое понимание структуры. Эта филигранная работа сделана Кириакосом Николау, которого часто неявно считают третьим в искусстве синтеза после Вудварда и Кори – более почётного и престижного третьего места представить трудно. И мы видим класс исследователя по этой с виду незаметной стадии. Проблема в том, что работать пришлось с очень чувствительной к условиям реакций системой трополона, которая, например, не выносит оснований даже достаточно мягких. И собран этот трополон (метиловый эфир) рядом с стерически нагруженным циклопентановым кольцом с тремя стереогенными центрами, но еще и с возможностью атропоизомерии. В такой трополон удалось воткнуть из галогенов только хлор (Br и I не полезли), поэтому еще и условия кросс-сочетания потребовалось подбирать под непростую задачу – и C-Cl ввести в окислительное присоединение, и трополон ненароком не раскурочить. Из классики кросс-сочетания годится только Судзуки-Мияура, но в варианте без основания – для этого придуман реагент, представляющий собой сразу анионный боронат, который прямо перед реакции делают в том же ведре из метокси-BBN и метиллития. Для нужной степени активации отлично подошел предкатализатор Бакуолда четвёртого поколения с лтгандом SPhos. Загрузка огромная, опять 50 мольных процентов, здесь не до экономии. Реакция получилась фактически стехиометрическая, хотя надо учесть еще и побочные подукты протодехлорирования, которые образуются из того же каталитического цикла, так что TON чуть больше единицы, TOF приблизительно 1 цикл за три часа. Реакция идёт с сохранением атропоизомерной конфигурации, но это я разберу в подробной части.
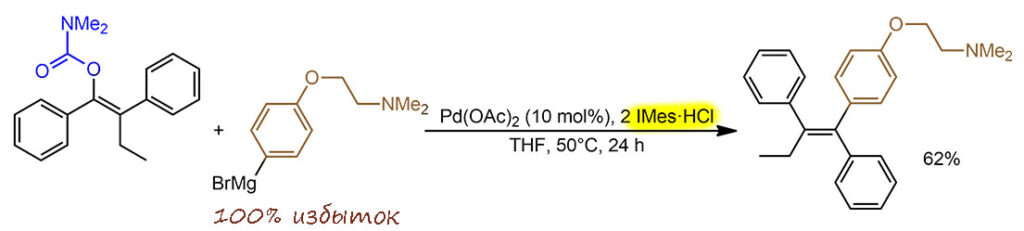
G-13
Реакция КТК, но с очень необычной уходящей группой, карбаматом. При выходе в 62% TON всего 6 циклов, TOF 1 цикл в 4 часа, или 0.25 цикла в час – очень медленно, но и реакция такая, и стерика мешает. Плохой выход еще усложняет выделение продукта, так как непрореагировавшие субстраты в больших количествах тоже надо быдет отделять. Активный анциллярный лиганд – сильнодонорный стерически затруднённый карбен IMes, обеспечивающий и активацию связи C-O в окислительном присоединении, и, скорее всего, и дальнейшую работу каталитической системы. Реакция диастереоспецифическая, идёт, как положено, с сохранением конфишурации. Авторы для перестраховки написали, что степень сохранения 99%, но это обычные предосторожности, так как это просто степень точности, достижимая при использовании ЯМР для оценки чистоты продукта.
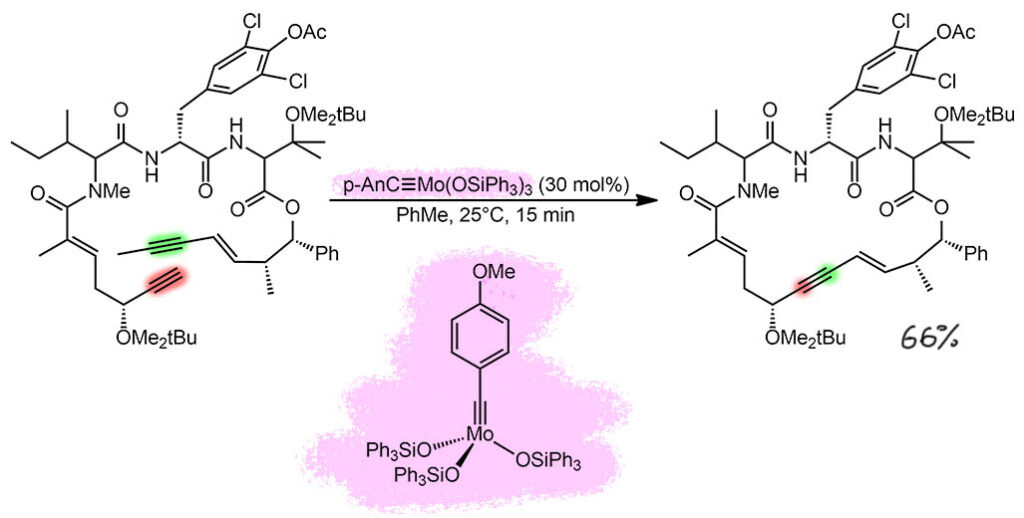
G-14
Метатезис алкинов (точнее, циклизация метатезисом алкинов – RCAM), еще один пример из работ Алоиза Фюрстнера, главного чемпиона (здесь английский смысл этого слова отлично резонирует с нашим) алкинового метатезиса, укротившего эту впечатляющую, но очень капризную реакцию, и давшего ей повестку явиться в большой синтез, где её уже давно заждались. Мы уже знакомы с семейством молибденовых катализаторов Фюрстнера, в которых силанольные лиганды удачно модулируют электрофильность молибдена и нуклеофильность карбина – получилась серия катализаторов, способных сшивать алкины (в основном в реакциях макроциклизации, а для алкинового метатезиса любая циклизация это макроциклизация) в молекула, содержащих множество функциональных групп с донорными атомами, которые с оригинальным алкиновым метатезисом совсем не работали, потому что блокировали молибден, насыщая его координационную сферу. В этой работе использовано очередное улучшение в этой серии катализаторов, точнее предкатализаторов, потому что и здесь есть обычная проблема – как стабилизирвоать комплекс для удобства и хранения (для этого нужны лиганды) и сделать его предактивацию в реакционной смеси наиболее легко и быстрой (для этого вспомогательные лиганды должны легко диссоциировать). Этого удалось добиться довольно просто с помощью немного более донорного карбина с пара-метокси-группой. Результат впечатляет, как в этом примере идет очень быстрая сшивка. Здесь есть еще одна фишка, обнаруженная Фюрстнером и его коллегами: мы уже знаем, что для алкинового метатезиса не годятся терминальные алкины, поэтому обычно берут с одной стороны метилы, и это дает второй продукт бутин-2, который мешает, потому что не так уж и летуч. Но оказалось, что вполне можно с одной стороны взять терминальный алкин, тогда образуется метилацетилен, намного более летучий. Правда здесь реакция идет 15 минут, так что не сполне ясно, успевает ли он улетать. Так или иначе выход хороший, а для макроциклизации просто великолепный. Загрузка большая, это довольно типичный прием, позволяющий сократить время реакции, и тем самым отсечь более медленные побочные реакции. Для метатезиса, в котором нет явно выраженных каталитических циклов, TON/TOF формальны, но здесь TON 2 цикла, и TOF – 8 циклов в час.
G-15
CH-арилирование, катализируемое комплексом Ru(2+). Как положено, это направленная реакция, направляющей группой свляется остаток тетразола, точнее, ближайший атом азота, обеспечивающий направленное металлирование (рутенирование). Направляющий азот именно этот даже в продукте двойного арилирования, не забываем, что эта штука крутится – воткнула арил с одной стороны, повернулась и воткнула с другой. В реакциях этого типа именно это основная проблема, потому что нужен обычно продукт моноарилирования, а бис-арилированный это побочный продукт. Оптимизация включала простой анциллярный лиганд, трифенилфосфин, и очень необычную добавку калийной соли сульфокислоты, но это мы разберем в подробном разборе. Если учесть побочное и то, что на него потрачено два цикла. то в реакции весьма почтенный TON под 100 циклов, впрочем и температура реакции недетская. TOF около 15 циклов в час. Предкатализатор – типичнейший для этой химии димерный ареновый комплекс с цимолом.
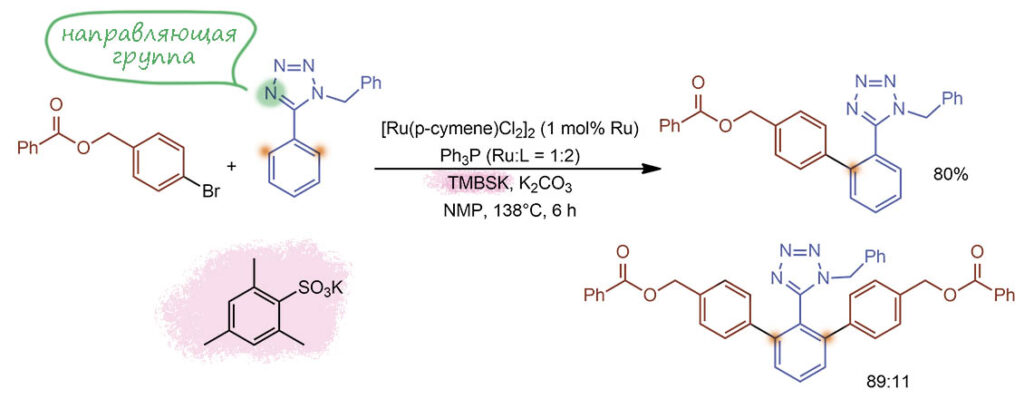
G-16
Реакция Негиси в самом чо ни на есть основном варианте – чистый классический предкатализатор без добавок, в ТГФ при небольшом нагревании, а цинкорганика приготовлена прямо перед добавлением палладия в том же ведре. Выход скромный, фактически реакция кажется некаталитичекой, подозрительно почти совпадают загрузка и выход, но не надо забывать, что здесь не одна, а 4 последовательные реакции, поэтому здесь TON 4 цикла. И мы еще понимаем, что каждый следующий остаток влезает тяжелее предыдущего, потому что все очень объёмистое и стерика нарастает очень серьёзно. Поэтому наша оценка TOF в 4 цикла за 72 часа, 1 цикл за 18 часов, явно просто формальность, потому что скорее всего первый остаток влетел ласточкой, а последний как раз и вколачивали львиную долю времени. Но чтобы это узнать, надо было пихать в молекулу остатки по одному, каждый раз выделяя промежуточный продукт и замеряя время реакции. Понятно, что это никому не нужно вообще. Нужен этот интересный продукт, который вполне мог вообще не получится, потому что это просто очень напряженная молекула с априори непонятной структурой.
G-17
Выход 43%. В общем, это вариация на тему Хека, только элиминирование гидрида с другой стороны. Анцилляр – трифурилфосфин, роль лиганда – чисто держать палладий, чтобы не пачкал колбу, все остальное анцилляра не требует или если и требует (гидридное элиминирование может хорошим лигандом ускориться), но для этой реакции проявляется типичное “противоречие Хека” – хороший анцилляр занимает слишком много места в координационной сфере, и до последней стадии дело может просто не дойти. Поэтому удовлетворяются этими простыми и слабыми лигандами, которые немного страхуют Pd(0), но больше скорее не мешают, чем помогают. TON маленький, 4 цикла. TOF медленный – цикл в три часа, треть цикла в час. Но мы уже насмотрелись на реакции гидридного элиминирования с образованием алленов и знаем, что они тугие и медленные. А низкий TON как раз и говорит о малой живучести катализатора, обслуживаемого слабым лигандом.
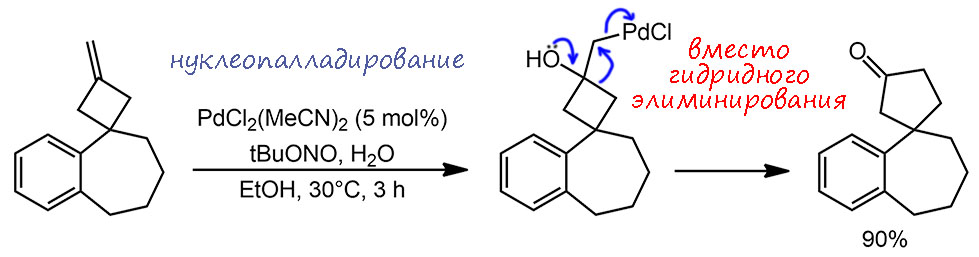
G-18
Реакция Вакер-Цудзи, но с необычным завершением в виде перегруппировки карбокатионного типа с расширением кольца, что можно расценить как осложнение, хотя с другой стороны, весь процесс можно считать новой палладий-катализируемой реакцией, требующей расширения стандартного набора простых реакций на возможность ухода Pd(2+) с лигандами в виде нуклеофугной группы, сопровождаемого восстановлением палладия до Pd(0) не только от аллильной системы, но и от насыщенного атома углерода. Процессы такого рода до этой работы не наблюдались, но здесь придумали создать очень сильную движущую силу в виде выгодной перегруппировки циклобутанольной системы с расширением цикла – высвобождение энергии напряжения очевидно и движет систему по этому пути. Катализатор в этом случае простой ацетонитрильный комплекс дихлорида палладия, а каталитическая система обеспечивается регенерацией необычным окислителем, трет-бутилнитритом. TON 18 циклов, TOF 6 циклов в час.
G-19
Тандем (ортогональный) реакций внутримолекулярного эндо-диг-гидроксиаурирования и кислотно катализируемой спироциклизации. Промежуточный продукт не выделяли. По конечному продукту выход 69%, и это дает нижнюю границу оценки TON в 7 циклов. То же с TOF, мы не знаем длительности второй стадии спироциклизации, поэтому приходится этим пренебречь и оценить TOF приблизительно в 10 циклов в час, очень неплохо. Реакция нуклеометаллирования, катализ катионным комплексом золота(1+), анциллярный лиганд опять JohnPhos. Реакция внутримолекулярного эндо-диг-гидроксиаурирования, анти-диастереоспецифическая, ненаправленная. Отличный пример совершенно выдающейся хемо- и региоселективности реакций, катализируемых золотом – посмотрите сколько всего в этих молекулах, а реакция действует как лазерный скальпель микрохирурга. И подумать только – золото это очень недавний пришелец в органическом синтезе, это химия этого века и этого тысячелетия.
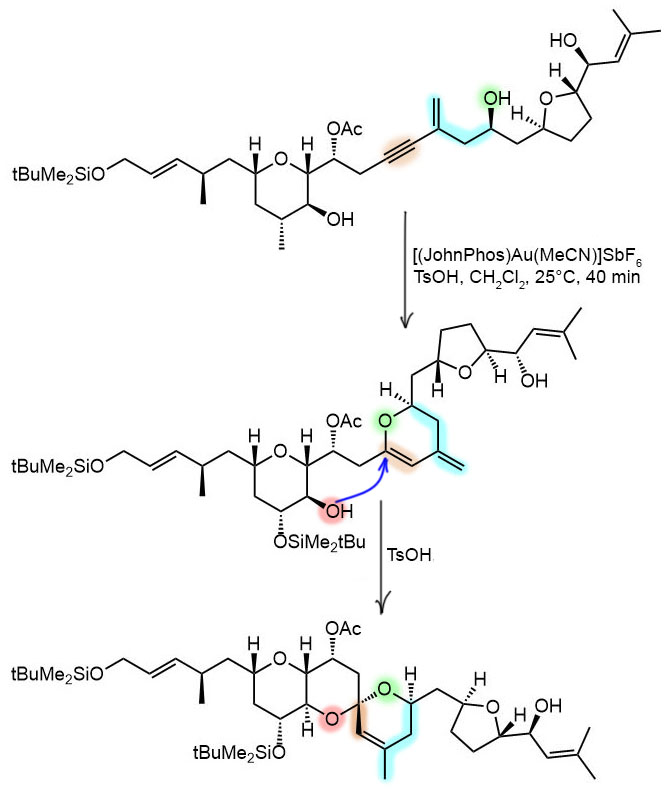
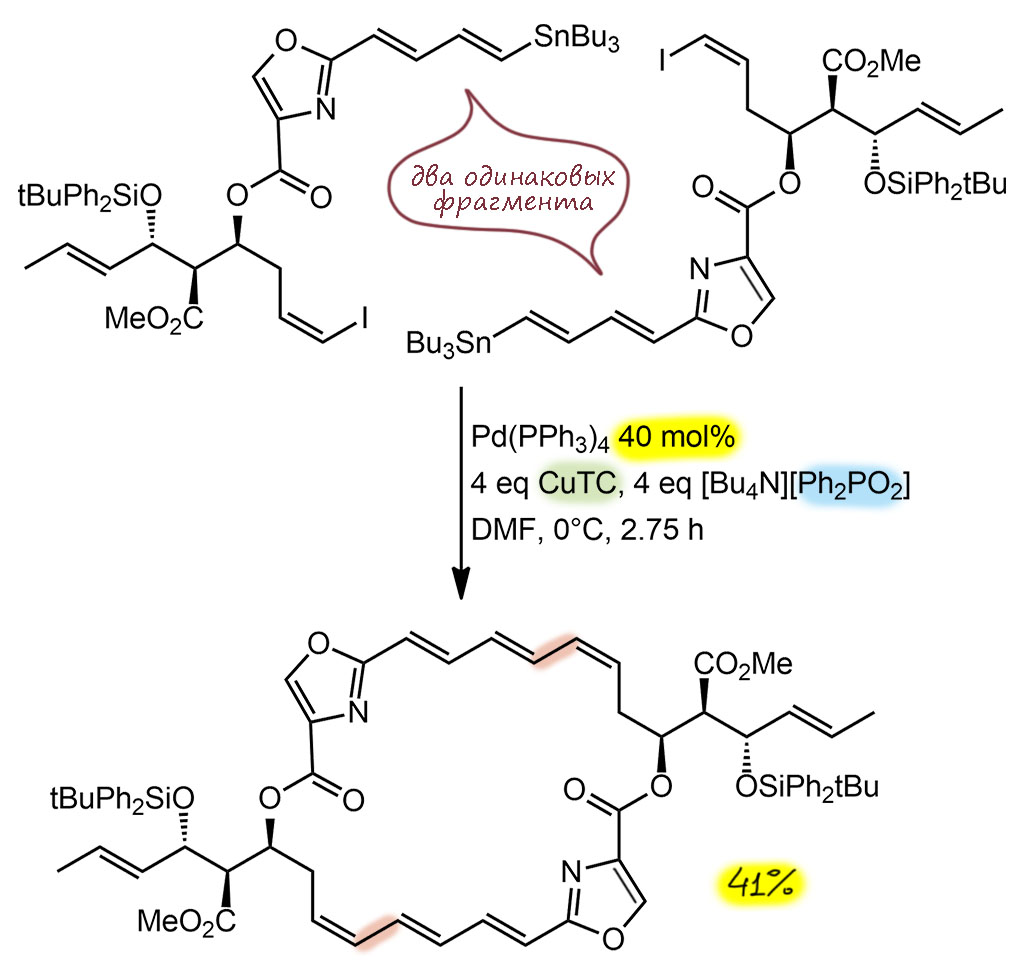
G-20
Кросс-сочетание по Стилле, почти классический вариант, добавка хлорида лития очень часто используется с самого начала, считается, что это помогает переметаллированию, немного активируя олово за счет гипервалентной координации. Предкатализатор стандартный комплекс Pd(0), анциллярный лиганд трифенилфосфин. TON опять небольшой, при загрузке в 10 моль% и выходе 64% это чуть больше 6 циклов. Еще раз убеждаемся что в сложных синтезах, да еще и почти в конце никто не занимается выжиманием побольше циклов. Выход правда скромный, но это потому что получился не сам продукт кросс-сочетания, а продукт последующей реакции, скорее всего, это внутримолекулярное аллильное замещение по Цудзи-Тросту,см. ниже. Если так, то это типичный тандем (авто-тандем) – активная форма катализатора после Стилле находит еще один реакционный центр и производит там некоторое действие. Естественно, потери могут быть именно на этой стадии, поэтому у самого кросс-сочтетания TON мог быть больше, но точно не больше 10, поэтому это непринципиально. TON – 6 циклов за 18 часов, 1 цикл за три часа, одна третья цикла в час – медленно. Вот, говорили-говорили, что кросс-сочетание такая потрясающая реакция, чисто-быстро. А здесь ни чисто, ни быстро, а задача-то всего-навсего винильный трифлат и первичное алкилолово. Но и нагревание нужно, и реакция идет медленно. Зато в молекуле много активных реакционных центров – кетон, даже енон, свободный гидроксил, эпоксид, третичный хлор. Но с гидроксилом что-то все же произошло и потребовалась защита, см. ниже, но все остальное нетронуто и не помешало. Это – очень сильное свойство кросс-сочетания. Ни енон, ни эпоксид не защитишь, и если бы такого метода не было, пришлось бы откладывать эти фрагменты на попозже. Все остальное нерелевантно – нет здесь ни стереоселективности, ни направленности. Есть осложнение.
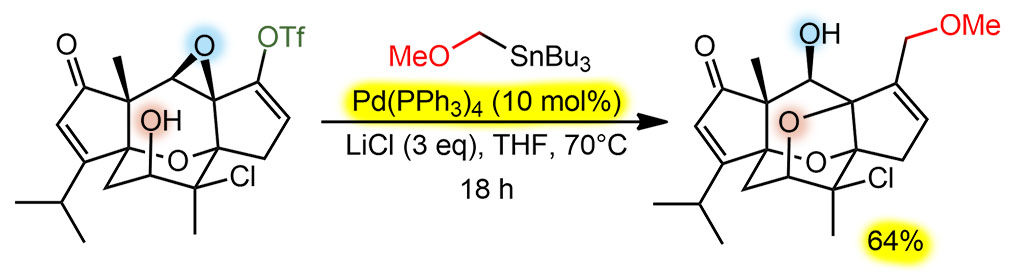
G-21
Реакция сначала кажется очень странной, но ничего особенного в ней нет. Это самый обычный внутримолекулярный Хек с гидридным элиминированием на соседний атом со смещением двойной связи. Продукт такого Хека был бы очевиден. Но в этом продукте сложилась система, очень хорошо приспособленная для гомоаллильного δ-элиминирования с образованием циклопропанового кольца под действием основания, которое и так присутствует для того чтобы обеспечивать катализ реакции Хека. Судя по тому, что сам продукт Хека до элиминирования вообще выделен не был, это очень быстрая реакция, видимо, потому что жёсткое сочленение колец создает наиболее удачные условия для орбитальных взаимодействий в переходном состоянии. Итак, оцениваем TON – даже до 2 циклов не дотягивает. Это, конечно, смешно звучит – раз не дотягивает, значит где-то на полдороге остановился? Все нормально, и ясно говорит нам, что реакция принципиально имеет каталитическую природу, а большую загрузку взяли только чтобы на одной из поздних стадий синтеза не рисковать, а получить один из ключевых для замысла фрагментов – три кольца на одной связи, так называемый пропеллан. TOF, понятно, тоже невелик – грубо по 10 часов на цикл.
G-22
Весьма типичная энантиоселективная реакция Хека. В таких реакциях стереогенный центр всегда создается за счет гидридного элиминирования на соседний атом, то есть с миграцией двойной связи. И здесь не исключение, но есть несколько интересных особенностей этой работы. Во-первых, карбопалладирование идет на пятичленный цикл индола, то есть на ароматическое кольцо, и такие реакции часто считают не Хеком, а СН-арилированием. Но здесь CH-активация исключена, потому что все положения замещены, и всё что остаётся, это карбопалладировать, что оказывается вполне возможно и создаёт спиро-сочленение, а также убирает ароматичность с этого кольца (модный нынче термин деароматизация – лишение ароматичности, а вы что подумали?). В этой стадии происходит перенос хиральности. Вторая фишка – обычно для асимметрической индукции в Хеке берут бидентатные хиральные лиганды и тогда приходится брать трифлат из-за дефицита координационных мест. А здесь взяли бромпроизводные, и пришлось подбирать монодентатный фосфин, в качестве которого опять хорошо зашли фосфоримидиты, уже разобранные нами в другом задании. Одна вещь здесь странна – чисто экспериментально авторы нашли, что результаты получаются лучше, если в смесь добавить муравьиную кислоту, причем никаких гидридных переносов не наблюдается. Понять роль этой добавки очень трудно, и мы погадаем еще в полном разборе. Итого – реакция внутримолекулярная, экзо-триг, энантиоселективная с очень хорошим оптическим выходом в 96%. TON приличный, 18 циклов. Но медленная, и это неудивительно, здесь и стерика тяжёлая (впрочем она же помогает энантиоселективности), и бром, а температура невысока, и лиганд в общем довольно поганый, если забыть об энантиоселективности, и подумать об окислительном присоединении. Но хорошая энантиоселективность оправдывает всё.
G-23
Энантиоселективная реакция Судзуки-Мияуры. Есть не очень много способов сделать кросс-сочетание энантиоселективной, поскольку этот тип реакции, по крайней мере, в рамках классики, в основном применим к ненасыщенным субстратам, а что-то сделать со стереогенными sp3-центрами практически невозможно. Можно сказать, ситуация отчаянная. Но к сачстью, есть другие элементы хиральности кроме стереогенных центров – очь и плоскость, и даже спираль. И вот эти штуки небезнадёжны. Энантиоселективность, связанная с атропоизомерией, уже давно разрабатывается в кросс-сочетании, но это делается на соединениях двух колец, которые цепляются друг за друга. А в этой работе решили вместо одного кольца взять просто непредельный остаток. Почему это так непросто и интересно, разберём в подробной части, но здесь затея удалась. Реакцию пришлось делать в мягких условиях, потому что атропоизомерные энантиомеры этого типа очень легко рацемизуются. Лиганд опять из БИНОЛа (ух, надоел уже, постылый), но хотя бы не гидрированного, и не фосфоримидит, а самый обычный фосфит. Вообще лиганд очень простой, даже удивительно простой. Хиральный безусловно, но без обычных обвесов, обеспечивающих стерические препятствия и значительную разницу в подходах к прохиральному элементу. Но всё получилось, а когда все получается, то чем проще, тем более лучше. TON около 19 циклов, TOF приблизительно 1 цикл в 3 часа. Оптическая чистота для такого сложного случая великолепна, ee 92% при высоком материальном выходе. Это интересная работа, доволньо здорово раздвинающая границы наших представлений об атропоизомерии и связанной с ней хиральности. В подробном разборе я даже кое-какие расчеты сделал, чтобы убедиться в том, что вообще такое возможно.
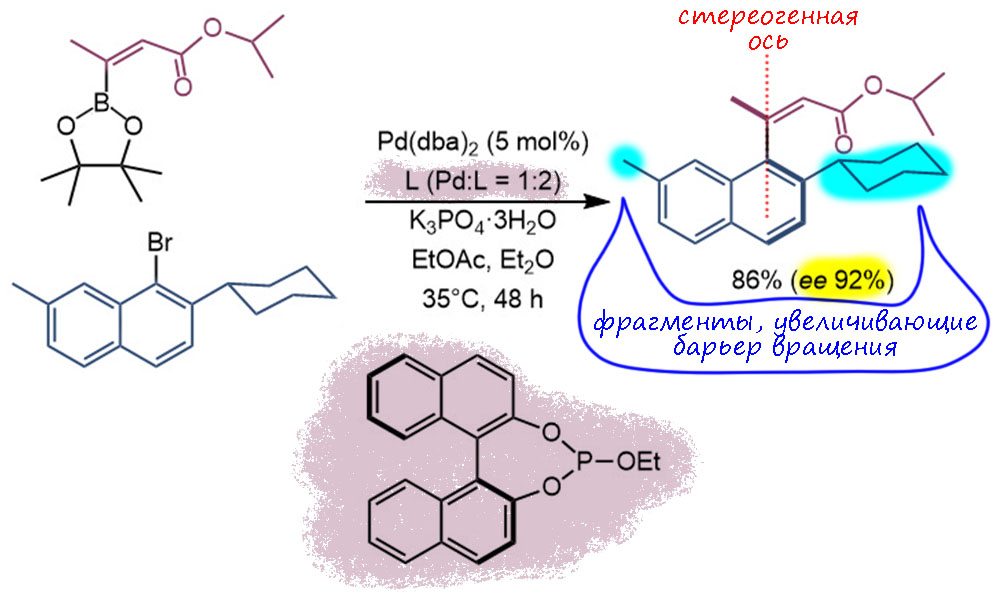
G-24
Реакция Бакуолда-Хартвига с новым протоколом, специально разработанным для сочетания с галогенпроизводными пятичленных гетероциклов. Используется новый анциллярный лиганд GPhos из семейства XPhos-BrettPhos, гемилабильный донорный монофосфин, в виде предкатализатора 6-го поколения типа комплекса окислительного присоединения. TON невелик, 16 циклов. TOF получше, в час пролетает 5 циклов с горкой, на цикл приходится всего 20 минут, и для C-N кросс-сочетания это очень хороший результат, показывающий потенциал каталитической системы, хотя реакцию ведут при нагревании и немаленьком – повышенная температура, увы, способствует деградации катализатора и невысокой живучести, что, скорее всего, и отражает невысокий TON. Реакция нестереоселективная, но имеющиеся стереоцентры не затрагиваются (иногда это немного лукаво считают высокой энантиоселективностью, ведь могло рацемизоваться? – могло! – а не рацемизовалось, вот! – значит высокая). Впрочем, всё равно отметим, что заслуга в этом может быть и в использовании мягкого основания. Реакция не направленная, межмолекулярная.
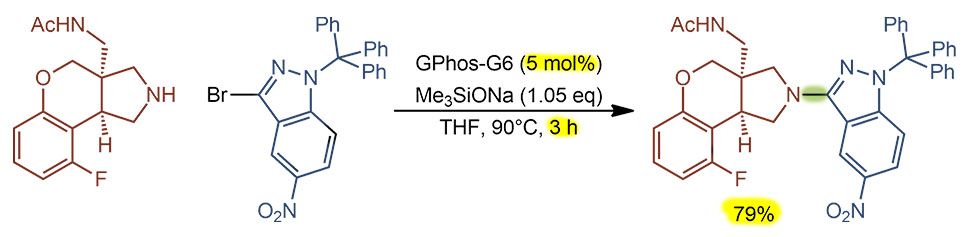
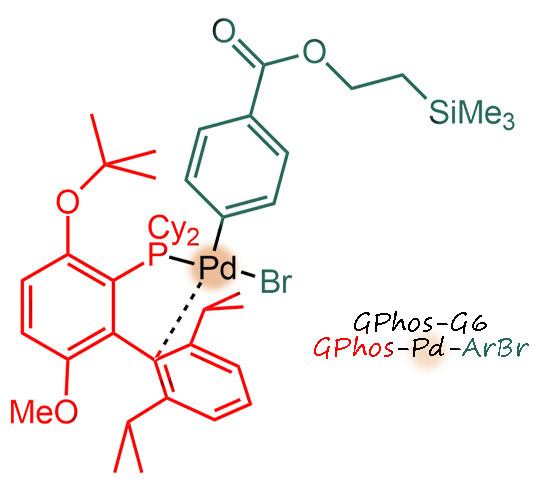
H-01
Последовательность реакций. Сначала CH-борилирование ароматического соединения, коронена. Реакция эта не такая селективная, потому что в основном, как мы знаем, управляется стерикой, и очень необычным и неоднозначным способом. В этой работе, судя по дополнительной информации, получилась смесь три и тетраборилированных, из которой хроматографией удалось выделить самое симметричное производное. Попробуем прикинуть TON, хотя это непросто. Во-первых, обращаем внимание на то, что загрузка иридиевого предкатализатора не учитыват то, что это димер, а если пересчитать, то получим 20 моль% иридия. При выходе в 21% кажется, что это некаталитическая реакция. Но борильных остатка три, и только это дает три каталитических цикла минимум, при этом мы понимаем, что циклов было больше и потому что не учтены другие продукты борилирования, и потому что 21% конечного продукта ничего не говорит о том, сколько было моно и диборилированного, ведь третье борилирование вряд ли количественно. Поскольку ничего этого мы не знаем, что просто пишем, что TON не менее 3. Соответственно TOF 1 цикл (то есть одна борильная группа) за 8 часов. Медленно, но верно.
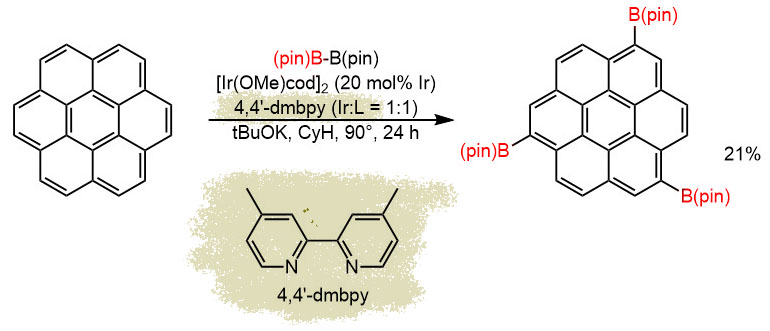
Вторая стадия это уже не простая реакция, а что – каскад, тандем или “в одном ведре”? Там очевидно все начинается с реакции Судзуки-Мияуры, которая очевидно доходит до конца, завершая свой цикл. Значит это точно не каскад. При этом образовалась каталитически активная частица, которая может немедленно пойти на второе окислительное присоединение. Где? Ведь мы не знаем, что происходит и в какой последовательности – может сначала все три Судзуки-Мияуры и только затем CH-арилирование. Или сначала на одном остатке все, а потом на другом, а потом на третьем. Или вообще случайно. Покольку PdL всегда уходит в раствор и за дело берется по-новой, у нас нет никаких ключей для разрешения этого вопроса. Значит, каждая реакция на каждом атоме бром это отдельный каталитический цикл, это не каскад. И раз одна реакция не доходит до конца (например, все три СМ) прежде чем начнется следующая, так что можно было бы при желании выделить промежуточный продукт и ввести его в следующую реакцию, значит это не “в одном ведре”. Следовательно это тандем, тандем Судзуки-Мияуры и CH-арилирования. Обратим внимание на страшно жесткие условия – аж 180°С! Это зачем и где такое видано? И растворитель какой-то странный, хлорнафталин. Это что такое? Какая из двух реакций требует такого? Ответ простой – никакая сама по себе. Это условия, которые нужны для того, чтобы эти огромные поликонденсированные молекулы оставались в растворе, а не выпадали безжизненной многослойной фанерой на дно из-за страшной склонности таких больших ароматических молекул к межмолекулярному взаимодействию по механизму пи-стекинга. Противостоять этому можно только ароматическим растворителем, который сам вмешивается в стекинг, разрывая взаимодейтсвие между отдельными поликонденсированными молекулами, но и то только при сильно повышенной температуре; а хлорнафталин взяли потому что он высоко кипит, и можно было вести реакцию при 180°С в обычной посуде без опасения, что ее разорвет в клочья. Единственное что странно, это то, что авторы не опасались кросс-сочетания борилированного коронена с хлорнафталином, ведь лиганд использовали сильнодонорный, вполне способный на активацию палладия для окислительного присоединения к C-Cl. Возможно, там это и получается, но желаемый продукт настолько сильно отличается от всех побочных и неполных продуктов, что выделяется легко и отделяется от всего остального, а у таких работ всегда бывает только одна цель – получить задуманное соединение, а какой ценой и с каким выходом неважно. Поэтому нет смысла здесь считать TON/TOF, тем более что с загрузкой предкатализатора в работе что-то странное. Во-первых, опять забыли про то, что в дибензилиденацетоновом комплексе два атома палладия. И если сделать на это поправку получается необычная история, что лиганда в два раза меньше, чем палладия. На деле это не так важно, потому что в этом комплексе содержание палладия не вполне соответствует формуле, а валентное состояние палладия переменно.
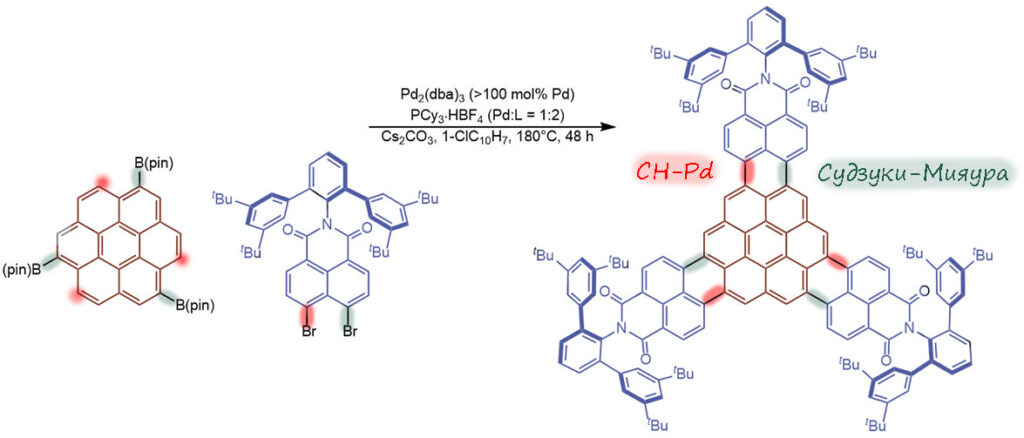
H-02
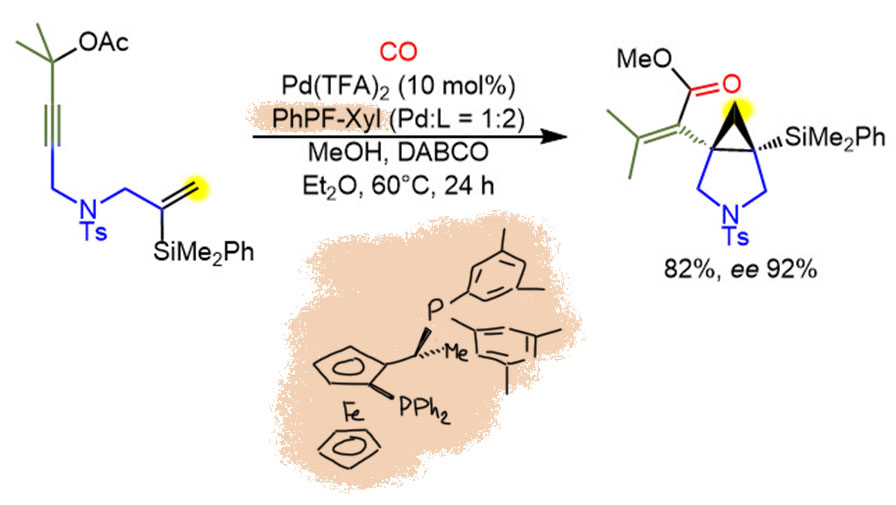
Это настоящий каскад, но с первой не совсем стандартной стадией формально окислительного присоединения, но с пропаргильно-алленильной перегруппировкой. Далее идут два карбопалладирования, первое экзо-триг с образованием 5-членного цикла, второе экзо-триг с образованием трехчленного цикла. Завершает каскад метоксикарбонилирование, которое, видимо, и тормозит весь цикл из-за бидентатного лиганда в координационной сфере, приходится поднимать температуру до 60 градусов, несмотря на то, что это наверняка ухудшает энатиоселективность. Опять компромисс. Анциллярный лиганд из семейства JosiPhos, является здесь и контролирующим лигандом каталитического цикла, и источником хиральности. Оптическая чистота ee 92%, неплохо, но не блестяще, повторю, видимо, потому что реакцию вели при повышенной температуре. Очевидно, что энантиодискриминация возникает на первом карбопалладировании, на втором результат уже стереоспецифичен, взаимное расположение задано, а карбопалладирование диастереоспецифично син. TON 8 циклов, как обычно в сложных реакциях, палладий (здесь в виде трифторацетата, веротяно, чтобы анион не мешал). TOF маленький: один цикл в 3 часа, 0.3 цикла в час. Предположительно, скорость ограничивает карбонилирование, и здесь еще и давление обычное, газ из надутого шарика, а при повышенной температуре растворимость газов (любых) уменьшается, так что и с концентрацией плохо. Можно было бы немного поднажать, хотя бы 2-3 атмосферы, но для этого нужно оборудование, и не получается ставить сто реакций в день, как нынче принято в местах, которые хотят угнаться за неумолимым темпом развития современной науки.
H-03
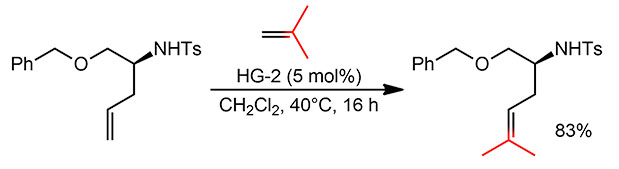
Это простая последовательность двух разных реакций кросс-метатезиса олефинов. Используется одинаковый предкатализатор – ховейда-граббс-2, у которого анцилляром является стандартный NHC SIMes. Хотя для метатезиса, строго говоря, TON/TOF не имеют большого смысла, потому что это не каталитический цикл, а разветвленная цепная реакция, да еще и с обратимыми стадиями, в которой комплекс металла является не столько катализатором, сколько инициатором и передатчиком цепей, формально прикинуть их несложно. Первая реакция TON пиблизительно 16, а TOF около 1 цикла в час.
Вторая – TON около 3 (трёх, да этот так, но так и задумано), а TOF – 1 цикл в 16 часов. Как мы выяснили в полном разборе, только так можно было получит кросс-метатезис с несимметричными и разветвленными олефинами и не использовать огромный избыток одного из олефинов.
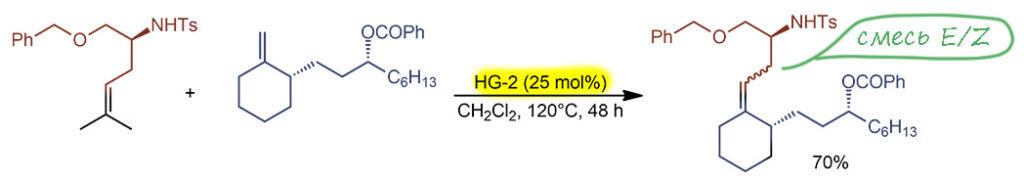
H-04
Это каскад окислительное присоединение-карбопалладирование-аллильное замещение, или Хек/Цудзи-Трост. С палладием здесь типичная заморочка – указана загрузка комплекса с двумя атомами палладия. Как это узнать? Придется лезть в дополнительные материалы и подметить то, что загрузка лиганда тоже указана в мольных процентах, и мы знаем соотношение палладий:лиганд = 1:2. Загрузка лиганда 20 моль%. Значит, палладия не пять, а 10 моль%, много, особенно учитывая, что перед нами не применение каскада в реальном сложном синтезе, а именно исследование методологии на моделях. Итого TON около 10 циклов, TOF приблизительно один цикл за 5 часов, или 0.2 цикла в час. Не быстро, но тут, как и в следующем примере, лиганд оптимизировали под высокий оптический выход, а его химическая активность, очевидно, невелика. Реакция энантиоселективна, хиральность переносится с хирального лиганда, о чём ниже, очевидно, на стадии внутримолекулярного аллильного замещения. Оптическая чистота довольно высока, 92%, и для этого подбирали температуру реакции, чтбы и выход был хорош, и энантиоселективность. остановились на компромиссных 40°С. Ниже оптическая чистота еще выше, но драматически падает скорость реакции и выход. Выше – уже быстро ухудшается энантиоселективность. Это обычный для энантиоселективных реакций компромисс. Направленности в реакции нет, нет смысла придумывать помощь окислителному присоединению от орто-гидроксила – там скорее не помощь, а помеха. Лиганд хиральный с двумя стереоцентрами, домашней сборки.
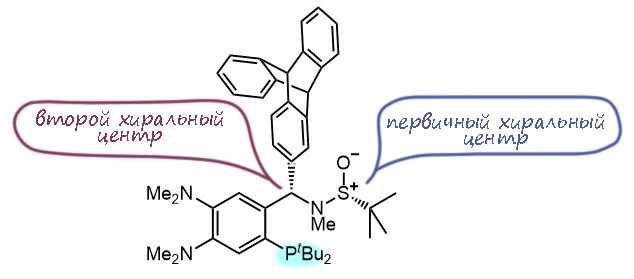
H-05
Каскад Кателлани практически в чистом виде, но норборнена мы не видим. Это не страшно. Когда каскад Кателлани стали применять к разным субстратам, выявились проблемы с оригинальным протоколом с участием норборнена. Проблема особенно остро проявляется, когда терминирующей реакцией является Хек – тогда возникает конкуренция карбопалладирования, обходящего каскад. Решение стали искать в замене норборнена, и как здесь, вышли на циклопентен, содержащий направляющую группу, который направляет палладий на положение рядом с полностью замещенным углеродом, что тоже эффективно блокирует гидридное элиминирование. С этим передатчиком каскада реакция фактически идёт как обычный каскад Кателлани. Более того, этот передатчик каскада позволяет селективно проводить алкилирование только с одной стороны. Поскольку это настоящий каскад, спокойно считаем TON – 18 циклов, TOF – 3 цикла в 4 часа, условно 0.75 цикла в час. Реакция Кателлани использует направляющие эффекты на стадиях внедрения передатчика каскада и циклопалладирования.
H-06
Это каскад, состоящий из окислительного присоединения, карбопалладирования, и терминированный реакцией Цудзи-Троста. TON невелик, около 20, но в реакции Цудзи-Троста это обычное дело, видимо, она и ограничивает здесь эффективность и живучесть каталитической системы. Поскольку реакция требует серьезного нагревания, для чего и взяли вместо обычного эфира более высококипящий МТБЭ. Повышенная температура реакции часто плохо сказывается на живучести катализатора, так как и всякие реакции деактивации тоже ускоряются, портится лиганд и не только. Реакция вдобавок еще и небыстрая даже при 80 градусах, и TOF – один цикл в 4 часа, или 0.25 цикла в час. Негусто. Опять обращаем внимание на проблему – никак не получается сделать так, чтобы анциллярный лиганд решал все проблемы, и высокую региоселективность, и скорость (TOF), и живучесть (TON), и энантиоселективность. Здесь всё неплохо с энатиоселективностью у основного продукта, ee 96%, при этом мы ничего не знаем про оптическую чистоту и конфигурацию побочных продуктов, а они вовсе не обязаны повторять характеристики основного продукта.
H-07
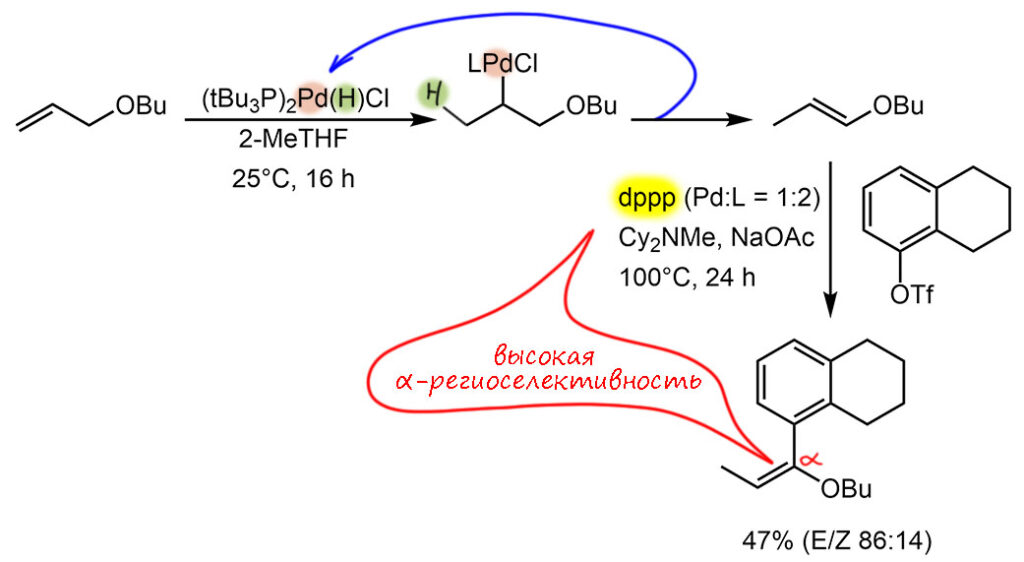
Немного странная работа, из серии “что бы сделать, если ничего более интересного в голову не приходит”. Естественно, в таких случаях на помощь приходят модные слова типа региодивергентности. В чём тут пресловутая дивергентность разберёмся в подробном разборе. А так авторы решили продемонстрировать, что виниловые эфиры можно не брать сразу, а генерировать их в реакционной смеси за счет палладий катализируемой изомеризации из намного более доступных аллильных эфиров, что чистая правда. Но правда и то, что аллилового эфира берут 5-кратный избыток, конечного продукта получают не более 50%, поэтому 450% исходного отправляется крокодилу под хвост. Так себе достижение, явно без перспектив в реальном синтезе, но не будем брюзжать, там довольно серьёзно взялись окучивать эту тему, может что приличное и вырастет. Для изомеризации они используют вполне стабильный готовый гидридный комплекс палладия с трис-трет-бутилфосфином. Реакцию проводят с 1 моль% предкатализатора, но выход мы не знаем и не узнаем, потому что исходный аллильный эфир берут в 5-кратном избытке относительно второго субстрата. Реакция потребовала 16-часовой экспозиции. Как и в многих реакциях Мидзороки-Хека с простыми субстратами, диастереоселективность оставляет желать чего-нибудь получше, образуется смесь E и Z (удивительно популярна эта буква в химии, ничего не поделать), получается немного комический эффект – региодивергентность есть, региоселективность даже есть, а стереоселективность плохая. Некоторые скажут – но этот виниловый эфир наверняка сразу же превратят в карбонильное соединение, и проблема с E/Z отпадёт. Наверное, хотя авторы работы особо упирают и на то, что продукт сохраняет структуру винильного эфира, а значит имеют на него другие планы.
Дальше в смесь добавляют арилтрифлат и типичные основания для варианта реакции Хека, давно разработанной для арилирования виниловых эфиров по α-положению, но для этого нужен еще и хелатирующий фосфин dppp. Его и добавляют, явно надеясь на то, что бидентатный фосфин вытеснит монодентатный с палладия. Надежда, видимо, оправдывается, потому что продукт действительно получается α-арилированный виниловый эфир. Надо заметить, что трифлат здесь опорный реагент, его в 5 раз меньше чем было исходного аллильного эфира, и палладия уже становится 5 моль%. Здесь мы можем прикинуть и TON (приблизительно 10 циклов), и TOF (приблизительно полцикла в час).
Самое спорное здесь это квалификация процесса как тандемного, даже, по новой терминологии, assisted tandem – тандем с содействием, имеется в виду то, что экспериментатор смело лезет в колбу и дерзко изменяет состав каталитической системы для второй реакции, здесь, добавляя другой лиганд. Проблема в том, что к этому моменту первая реакция закончена, и ничего больше не происходит, пока не будут добавлены новые реагенты и лиганд, и повышена температура. На мой взгляд, это обычный процесс в одном ведре (one pot). Тандемы всё же должны быть самопроизвольны – закончилась одна реакция, начинается другая. В этом месте в классификации явно не всё гладко.
H-08
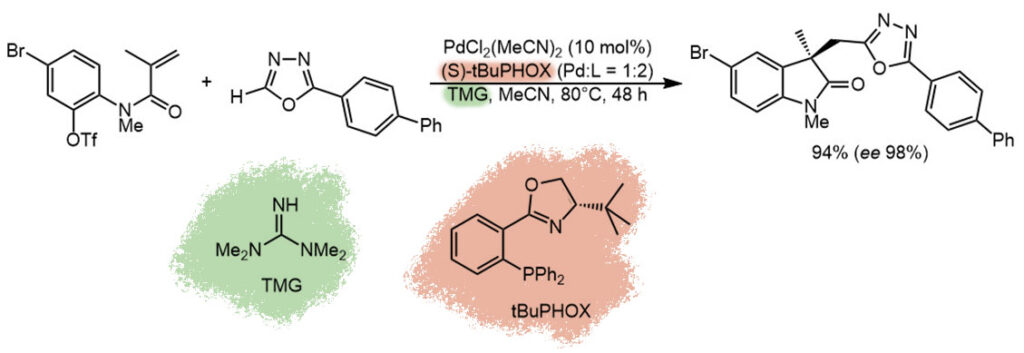
Это каскад окислительное присоединение – внутримолекулярное карбопалладирование – CH-арилирование. Карбопалладирование энантиоселективное. Анциллярный лиганд хиральный, он же источник хиральности, из семейства PHOX, комбинация фосфина и оксазолидина, хиральность обусловлена единственным стереогенным центром. Функционирование бидентатного лиганда в цикле, включающем карбопалладирование, обеспечивается трифлатом, не занимающим места в координационной сфере. TON невелик, около 9 циклов, типично для каскадов. TOF совсем мизерный, один цикл в пять часов, 1\5-я цикла в час, реакция медленная. и это не удивительно, СН-арилирования редко бывают быстрыми, эта стадия точно тормозит цикл. Последняя стадия цикла ненаправленная, межмолекулярная, хотя можно представить и предварительную сборку реакционного комплекса за счет взаимодействия палладия с гетероатомами в гетероцикле, но увы, для этого у палладия не хватает координационных мест.
H-09
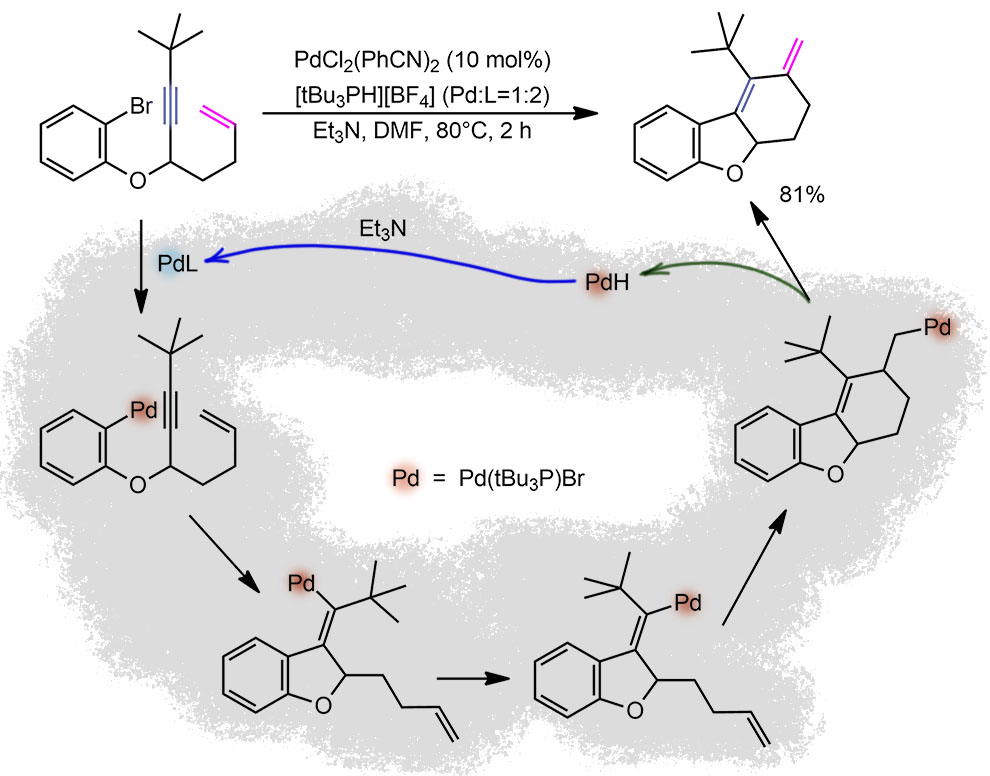
Это каскад, включающий не совсем обычную стадию изомеризации интермедиата карбопалладирования. Каскад всключает стадии окислительного присоединения – экзо-диг-карбопалладирования – син-анти-изомеризации – и терминируется внутримолекулярной экзо-триг реакцией Мидзороки-Хека. Анциллярный лиганд – трис-трет-бутилфосфин, не вполне понятно за каким чёртом, ведь вся работа с другими примерами сделана с трифенилфосфином. Вместо мерзкого, токсичного и пирофорного фосфина взята его кристаллическая и устойчивая соль, которую можно взвешивать на обычных весах на воздухе, но этот трюк давным давно открыл Грегори Фу, один из главных поклонников этого фосфина в катализе. TON, как обычно в каскадах, невелик – 8 циклов, но TOF не так уж плох, 4 цикла в час, уж не для этого ли и взяли этот фосфин вместо трифенилфосфина, с которым реакции шли по 16 часов. В реакции наблюдается необычная диастереоселективность из-за описанной в статье изомеризации интермедиата карбопалладирования дизамещённой тройной связи.
H-10
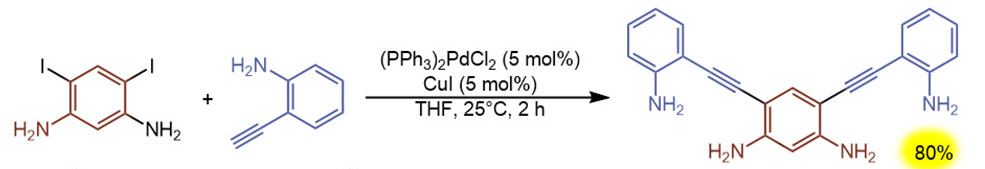
Простая последовательность двух реакций. Первая – Соногасира, в общем самая обычная, но трудно не заметить то, что в обоих субстратах есть орто-амино-группа, или даже две. И ничего – всё работает, протокол самый классический, основание – диизопропиламин, используется как сорастворитель в большом избытке, но реакция быстрая при комнатной температуре. Поскольку реакций две, TON 32 цикла, TOF – 18 циклов в час, тоже отлично, давно мы в кросс-сочетаниях такой прыти не видели. Безусловно, может закрасться подозрение, что амино-группы не только не мешают, а даже и помогают реакции, например, осуществляют направляющий эффект. Возможно, но это никак подтвердить сейчас не получится.
Вторая стадия – внутримолекулярное нуклеометаллирование с катализом комплексом золота, анциллярный лиганд – стандартный NHC IPr. В реакции могут участвовать амино-группы из среднего и крайних колец, во всех других примерах их статьи всегда идет циклизация на среднее кольцо, и только в этом на крайние с образованием двух индолильных остатков. И здесь две реакции, выход по совпадению тот же, поэтому и TON тоже 32 цикла, но TOF в два раза выше, 32 цикла в час, цикл в две минуты. Шустро. Циклизации эндо-диг.
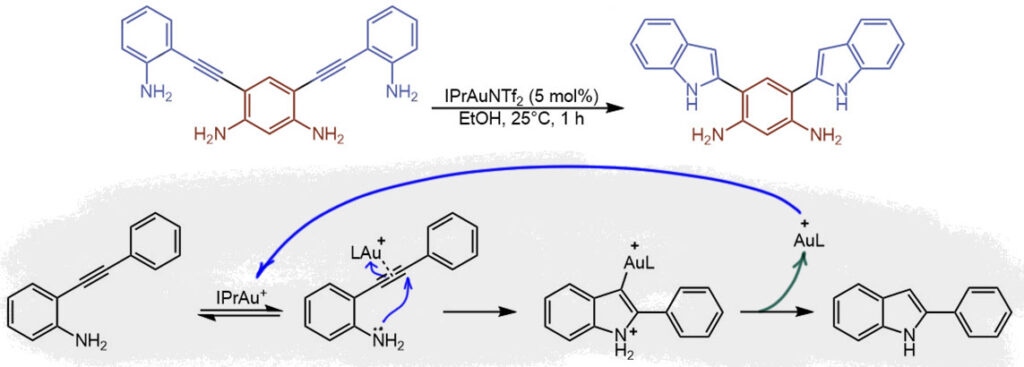
H-11
Последовательность из двух реакций. Первая – раскрытие цикла метатезисом с этиленом (ROM, ring opening metathesis, этенолиз). Чтобы реакция шла в этом направлении, используется довольно высокое давление этилена, иначе равновесие поедет в обратную сторону. Используется Ховейда-Граббс-2, причем половину от исходной загрузки удается после реакции достать из реакционной смеси. А почему не все 100%, это же катализатор?? Нет, это предкатализатор, напоминаю еще раз, что при предактивации состав каталитически активного комплекса изменяется, в частности, в нем меняется карбен. А почему тогда половину обратно достали? Скорее всего потому, что они и не вступали в процесс, предактивация далеко не количественная, а реакция метатезиса довольно быстрая. Можно также предположить, что этот комплекс регенерировался из стирола, получившегося из рутенациклической части предкатализатора, но это вряд ли происходит в существенной степени. А как же нам считать TON (уже не будем лишний раз напоминать, почему в метатезисе это формальность) – на полную загрузку или на половину? Да как хотите, так и считайте, ведь это все просто гипотезы, и с уверенностью мы не можем утверждать, что в реакции участвовала только половина. В общем, TON что-то между 8 и 16 циклами, а TOF от 2 до 4 циклов в час. При этом мы отлично понимаем, что большую часть времени рутениевые комплексы делают этилен из этилена, при таком избытке этого олефина, поэтому скорее всего комплексы работают намного быстрее, чем показывают эти формально-оценочные цифры, но вхолостую. Ничего не поделать, это метатезис.
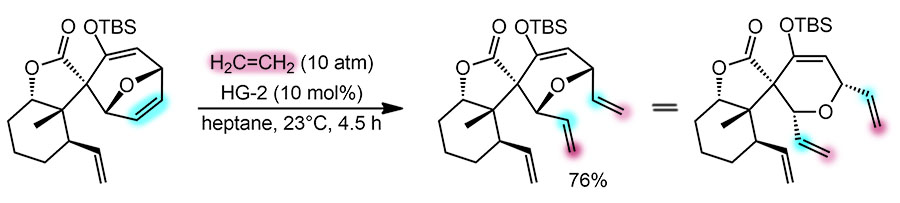 Вторая реакция это уже не так просто. Это очень любопытный каскад, в общем состоящий из карбопалладирований, но в начале довольно необычный – переметаллирование силилового эфира енола дает палладиевый енолят. Химия палладиевых енолятов известна давно, но используется нечасто, хотя это очень занятная химия. В подробной части разберемся с ней получше, а пока просто отметим, что палладиевый енолят (он же оксоаллильный комплекс) вступает в равновесие металлотропии O-енолята и C-енолята (и возможно, оксоаллильного комплекса), а уж С-енолят, являющийся самым обычным палладийорганическим интермедиатом, начинает последовательность двух карбопалладирований (обе реакции экзо-триг), но вторая заканчивается гидридным элиминированием, и это уже полноценный Хек – мы ведь договорились терминировать каскады не элементарными, а полными реакциями. Поэтому весь каскад можно расписать как переметаллирование – палладотропия – карбопалладирование – Хек. В реакции используется бесфосфиновый режим, только ацетат палладия как предкатализатор, и это потому что первая реакция с силиловым эфиром ингибируется лигандами (на самом деле, насколько я знаю, это просто никто не исследовал, можно ли там взять, например, нефосфиновый лиганд), поэтому используется стехиометрический режим (даже с небольшим избытком). Кроме того, пришлось бы рециклизовать Pd(2+) из гидридного комплекса. С этой проблемой и ее решениями мы сталкивались уже много раз, это требует поиска хорошего окислителя, который не будет мешать другим стадиям, и это весьма непросто, такой окислитель может захотеть реагировать с исходным силиловым эфиром (вот Cu(2+) точно захочет), и видимо, желающих упираться рогом ради небольшой экономии палладия пока не нашлось.
Вторая реакция это уже не так просто. Это очень любопытный каскад, в общем состоящий из карбопалладирований, но в начале довольно необычный – переметаллирование силилового эфира енола дает палладиевый енолят. Химия палладиевых енолятов известна давно, но используется нечасто, хотя это очень занятная химия. В подробной части разберемся с ней получше, а пока просто отметим, что палладиевый енолят (он же оксоаллильный комплекс) вступает в равновесие металлотропии O-енолята и C-енолята (и возможно, оксоаллильного комплекса), а уж С-енолят, являющийся самым обычным палладийорганическим интермедиатом, начинает последовательность двух карбопалладирований (обе реакции экзо-триг), но вторая заканчивается гидридным элиминированием, и это уже полноценный Хек – мы ведь договорились терминировать каскады не элементарными, а полными реакциями. Поэтому весь каскад можно расписать как переметаллирование – палладотропия – карбопалладирование – Хек. В реакции используется бесфосфиновый режим, только ацетат палладия как предкатализатор, и это потому что первая реакция с силиловым эфиром ингибируется лигандами (на самом деле, насколько я знаю, это просто никто не исследовал, можно ли там взять, например, нефосфиновый лиганд), поэтому используется стехиометрический режим (даже с небольшим избытком). Кроме того, пришлось бы рециклизовать Pd(2+) из гидридного комплекса. С этой проблемой и ее решениями мы сталкивались уже много раз, это требует поиска хорошего окислителя, который не будет мешать другим стадиям, и это весьма непросто, такой окислитель может захотеть реагировать с исходным силиловым эфиром (вот Cu(2+) точно захочет), и видимо, желающих упираться рогом ради небольшой экономии палладия пока не нашлось. 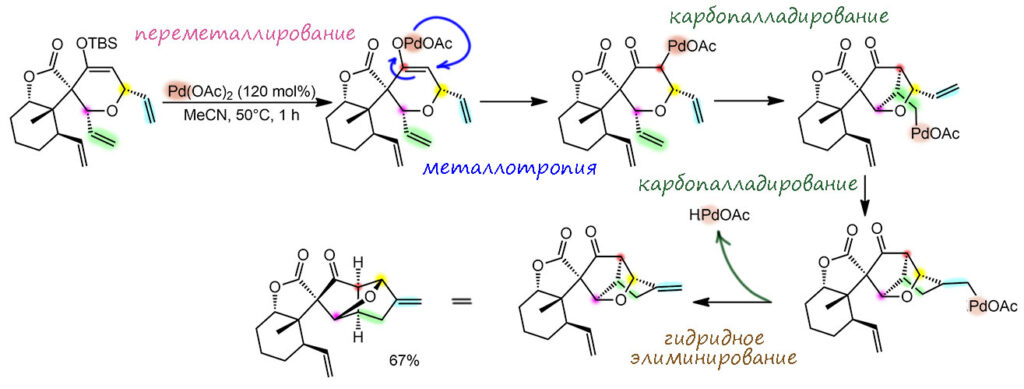
H-12
Довольно мутная работа, хотя внешне все выглядит очень привлекательно и просто: берут анилины, разные замещенные, и простую непредельную кислоту, винилуксусную (бут-3-еновую) и в условиях, похожих на Вакер-Цудзи, но с бидентатным лигандом, производным фенантролина, и необычным окислителем, трет-бутилгидропероксидом получают непростое соединение с тремя новыми циклами и довольно определенной стереохимией (в диастереомерном, естественно, смысле, никаких источников хиральности здесь нет), включающее две молекулы этой кислоты. Авторы считают, что это сложный каскад, начинающийся с азотистой версии Вакер-Цудзи ( это называют аза-Вакер), и далее двигающейся через внутримолекулярное направленное палладирование, карбопалладирование, просто Вакер-Цудзи, еще одно карбопалладирование, но связи C=N, и завершающееся обычным образованием лактама. Получается, что это даже не каскад, а тандем с участием каскада. Но, увы, обсуждение очень поверхностное, доказательства механизма неубедительные, и количество грубых швов с торчащими белыми нитками в работе удручает.
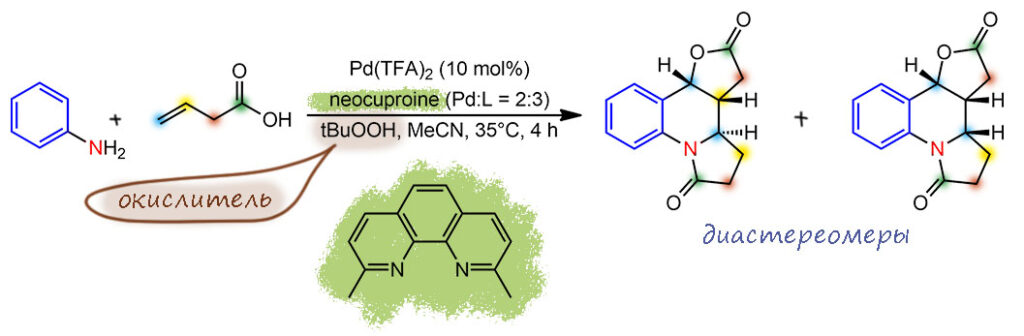
Поэтому я здесь воздержусь от повторения этого механизма, а в подробном разборе объясню, что здесь не так, и как можно было бы это поправить, хотя полностью убедительный механизм все равно не получается. Так бывает. И это довольно полезно – иногда разбирать работы, вызывающие больше вопросов, чем восхищения и ответов. Просто помечу на структуре задания, какие связи образованы, по мнению авторов работы, и с помощью каких реакций.

Поскольку как на это ни смотри, это не один каскад, то есть не один каталитический цикл, то считать TON здесь бесполезно. Оставим эту занятную, но возможно недопонятую реакцию до полного разбора.
H-13
Две отдельные последовательные реакции. Первая – борилирование по Мияуре, практически оригинальный протокол с комплексом палладия с dppf и ацетатом калия. TON – 7 циклов, и это за 20 часов, приблизительно 1 цикл в три часа или треть цикла в час. Величины скромные, но здесь явно нужен был продукт, а загрузки очень малыми, в таких случаях палладий не жалеют и никак не пытаются оптимизировать и минимизировать.
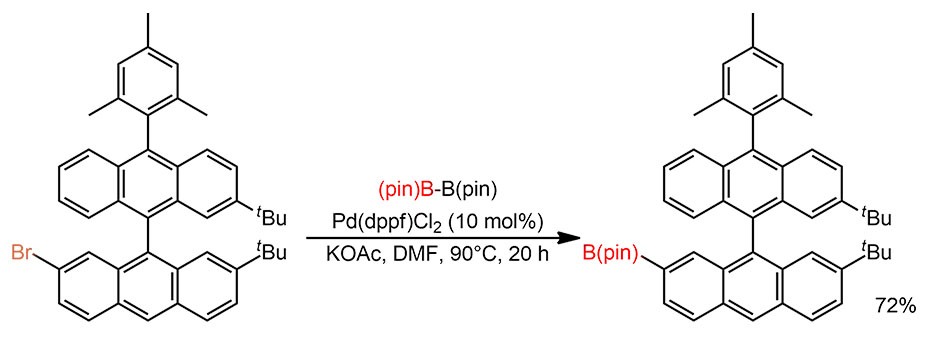
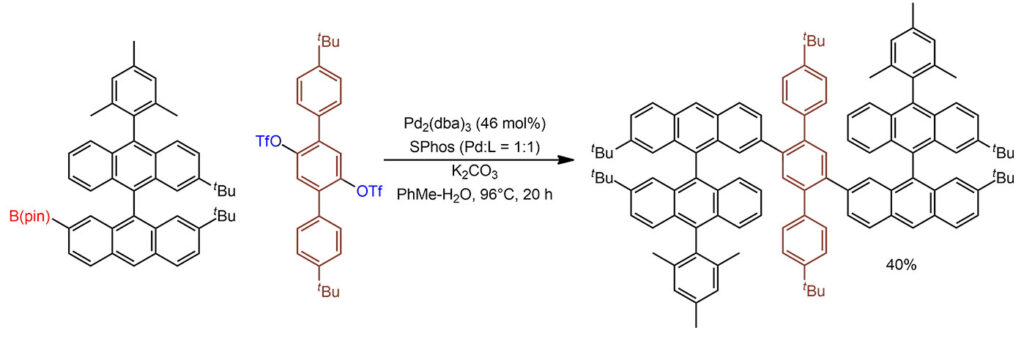
Вторая – Судзуки-Мияура с трифлатом. Главная проблема – очень большие молекулы с кучей конденсированных колец, с такими всегда бывают проблемы с растворимостью, поэтому используют ароматический растворитель и нагревание. Сложнее с загрузками, особенно с dba-комплексом. Судя по указанию в доп информации загрузки в весовых и мольных количествах, использовали сольват с хлороформом, видимо, еще и с поправкой на процентное содержане палладия. По непонятной причине загрузка палладия была соотнесена не с опорным реагентом (здесь трифлатом) с с борилированным, которое использовано с небольшим избытком по соотношению 2:1. Если прикинем еще и с учетом 2 атомов палладия, получим аж 46 моль% с соотношением к анциллярному лиганду, а здесь это SPhos 1:1. Так редко делают, когда берут простой предкатализатор, потому что невозможно достичь количественного связывания в равновесии, но здесь при такой большой загрузке предкатализаторв, всем просто никак не важны все эти TON. Мы все же прикинем, учитывая, что кросс-сочетания здесь два – и это 2 цикла, 1 цикл в 10 часов (TOF 0.1 цикла в час).
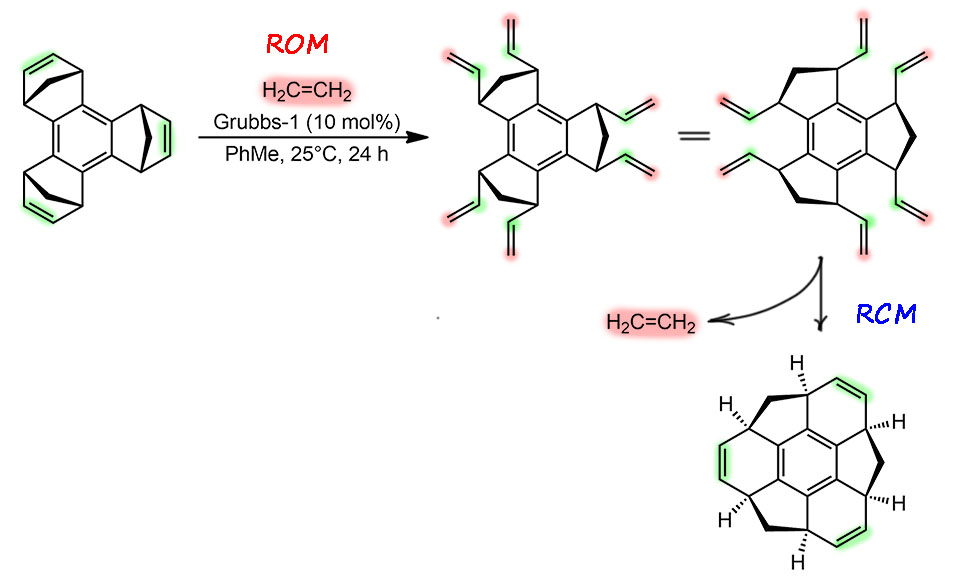
H-14
Очень похожий пример у нас был в прошлом году – метатезис сначала раскрывает циклы (ROM) реакцией с этиленом (этенолиз), а затем замыкает в другом порядке (RCM). Здесь это выглядит довольно красиво и используется в синтезе неплоского поликонденсированного углеводорода суманена. С метатезисом всегда проблема, потому что у него нет в полном смысле слова каталитического цикла, так как это цепная реакция. Здесь происходит раскрытие (этенолиз) цикла, а образовавшиеся винильные группы вступают в RCM, Хоть я и нарисовал сначала все три раскрытия (ROM), а затем три закрытия (RCM), но в реальности мы не можем гарантировать, что кольца не раскрываются и закрываются последовательно. Все реакции объединены участием простого карбенового (метилиденового) комплекса, поэтому их можно было бы смело считать каскадом. А может считать просто одной реакцией метатезиса, в которой участвуют разные промежуточные олефина и карбеновые комплексы. Реакция целенаправленно идет в одном направлении (хотя выход всего 30%, из чего следует, что значительная часть материала полимеризуется) по очень простой причине – это направление соответствует уменьшению напряжения, потому что бициклические структуры с мостиком превращаются в обычные 6-тичленные циклы. Формальная оценка TON/TOF может быть дана, если просто учесть, что произошло шесть отдельных реакций метатезиса (3 ROM + 3 RCM), и при загрузке предкатализатора Граббс-1 в 10 моль% и выходе 30% получается 18 циклов, и это за 24 часа, 3 цикла в 4 часа, один цикл чуть больше часа.