
Лиганды
Любое координационное соединение состоит из металла и лигандов. Атомов металла может быть несколько – тогда комплекс называется многоядерным или кластером. Мы почти не будем касаться многоядерных комплексов, ограничимся одноядерными, в которых атом металла один-единственный. Уточним, что речь у нас будет идти только о комплекcах переходных металлов. А у непереходных металлов бывают комплексы? Конечно, и выглядеть они иногда могут очень внушительно. Например, все знают такой знаменитый комплекс железа как ферроцен, FeCp2, называемый сандвичем потому что атом железа сидит между двумя ломтями циклопентадиена. У щелочноземельных элементов – бериллия, магния, кальция – тоже есть такие комплексы, металлоцены MCp2, и с виду их отличить от ферроцена очень трудно, просто близнецы. Только устроены они по-разному. У переходных металлов в образовании комплексов обязательно участвуют d-орбитали, образующие с орбиталями лигандов сложное многоцентровое взаимодействие. А у комплексов s-металлов связь между металлом и лигандами почти чисто ионная, части удерживаются вместе кулоновским притяжением зарядов.
Посмотрим на комплексы переходных металлов с простыми лигандами. Простые лиганды – это атомы или группы атомов, соединенные с металлом через один атом.
А как узнать, через сколько атомов соединен лиганд с металлом? Неужели просто “на глазок”? Почти, но не совсем. Нужно знать геометрию комплекса, то есть положение всех атомов комплекса в пространстве. Такую информацию дает рентгеноструктурный анализ, и данных по геометрии различных комплексов металлов накоплено огромное количество. Все они собраны в Кембриджской кристаллографической базе данных The Cambridge Crystallographic Data Centre (CCDC), откуда их можно по необходимости бесплатно выуживать. Зная геометрию комплекса, можно определить расстояния между атомом металла и окружающими атомами и найти те, которые расположены близко. Для каждой пары атомов известны характерные расстояния, и если наблюдаемое расстояние меньше такового, то это и считают признаком связи. Если расстояние существенно больше, то считается, что связи нет. Программы, предназначенные для показа структур на экране компьютера, например, Mercury, делают это автоматически и показывают обнаруженные связи, рисуя химическую структуру так, как мы к этому привыкли. На этом сайте я часто буду приводить структуры комплексов, взятые из реальных статей и базы CCDC, причем в основном буду использовать более наглядную графику, которую называют capped sticks – палочки с шапочками, палочки это связи, шапочки – это обозначения атомов окраской отрезков палочек, соответствующих атомам. Некоторые важные атомы, особенно атомы центрального металла буду для удобства показывать шариками. Отмечу, что в представлении рентгеноструктурных данных всегда считалось обязательным использовать проекции типа ORTEP (по названию программы, которая рисовала такие проекции по данным РСА), где атомы показываются эллипсоидами – по размерам этих эллипсоидов сразу видна степень неопределенности положения атома, а следовательно и качество структуры. Это очень важно видеть сразу, чтобы не пользоваться структурами низкого качества, а следовательно недостоверными. Но ORTEP проекции очень неудобны, если нам нужно просто как следует разглядеть собственно структуру комплекса. В последние лет 10 ORTEP медленно, но верно уступает позиции более наглядным проекциям, в которых атомы обозначаются шариками или просто цветом. Причина этого проста – сейчас очень легко вытащить любой оригинальный РСА-файл и проверить его на качество, а кроме того, современные требования к структурам таковы, что некачественные структуры просто не принимаются ни к публикации, ни к загрузке в CCDC.
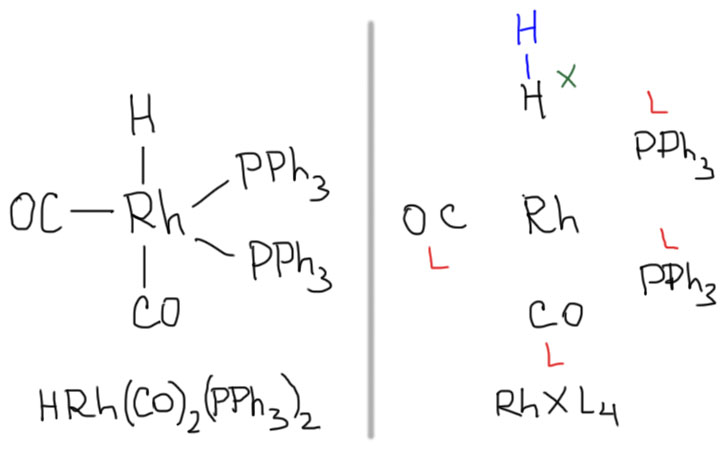 Итак, простые лиганды связаны с атомом металла через один атом. Простые лиганды удобно классифицировать на два типа: X-лиганды и L-лиганды. Возьмем какой-нибудь несложный комплекс и посмотрим, как он устроен. Слева нарисован комплекс родия с тремя разными лигандами. Кратко, в строчке, комплексы записываются достаточно произвольным образом, нет никаких требований на то, в каком порядке перечисляются лиганды, должен ли металл быть впереди или сзади и т.п. Справа тот же комплекс, разобранный на части: в середине металл, вокруг лиганды. Видим, что некоторые лиганды выглядят, как полноценные нейтральные молекулы: например, трифенилфосфин или оксид углерода. Пока оставим в стороне вопрос, как эти молекулы (лиганды) соединяются с металлом. Мы видим только то, что они связаны через один атом – фосфин через фосфор, а CO через углерод. Откуда видим, мы уже разобрались – кто-то до нас получил структуру этого комплекса, померил расстояния между атомами; нашел, что расстояния Rh-P и Rh-C достаточно малы, чтобы считать их соответствующими координационной связи, и нарисовал структурную формулу. Простые лиганды, которые выглядят как полноценные нейтральные молекулы, называются лигандами L-типа. Пока не будем заострять внимание, но отложим в памяти, что молекула не обязана быть стабильной, важно только чтобы она была нейтральной.
Итак, простые лиганды связаны с атомом металла через один атом. Простые лиганды удобно классифицировать на два типа: X-лиганды и L-лиганды. Возьмем какой-нибудь несложный комплекс и посмотрим, как он устроен. Слева нарисован комплекс родия с тремя разными лигандами. Кратко, в строчке, комплексы записываются достаточно произвольным образом, нет никаких требований на то, в каком порядке перечисляются лиганды, должен ли металл быть впереди или сзади и т.п. Справа тот же комплекс, разобранный на части: в середине металл, вокруг лиганды. Видим, что некоторые лиганды выглядят, как полноценные нейтральные молекулы: например, трифенилфосфин или оксид углерода. Пока оставим в стороне вопрос, как эти молекулы (лиганды) соединяются с металлом. Мы видим только то, что они связаны через один атом – фосфин через фосфор, а CO через углерод. Откуда видим, мы уже разобрались – кто-то до нас получил структуру этого комплекса, померил расстояния между атомами; нашел, что расстояния Rh-P и Rh-C достаточно малы, чтобы считать их соответствующими координационной связи, и нарисовал структурную формулу. Простые лиганды, которые выглядят как полноценные нейтральные молекулы, называются лигандами L-типа. Пока не будем заострять внимание, но отложим в памяти, что молекула не обязана быть стабильной, важно только чтобы она была нейтральной.
Один из лигандов – атом водорода. Атом водорода не является полноценной молекулой, это одновалентный кусок какой-то молекулы, например, молекулы водорода. Мы можем мысленно дорисовать недостающее. Если просто оторвать этот лиганд от металла, мы получим или радикал, или анион, или даже катион, но не полноценную молекулу. Такие лиганды принято называть лигандами X-типа. В таких обозначениях комплекс можно в общем виде записать как RhXL4. 
Металлоорганические соединения
Поскольку мы собираемся заниматься органической химией, а не просто химией координационных соединений, нам особенно будут интересны комплексы металлов с органическими лигандами. Из обычного курса органической химии мы знаем, что бывают металлоорганические соединения, и там мы часто встречаемся с литийорганикой и магнийорганикой, иногда с цинк-органикой. Всё это производные непереходных металлов. Всплывают там ещё и купраты, производные меди, а это уже переходный металл. В курсе органической химии никто не озаботится объяснить, что же такое металло-органическое соединение. Мы просто интуитивно причисляем к ним всё перечисленное и не паримся. А почему мы не считаем металлоорганикой, например, соли карбоновых кислот? А еноляты? Или считаем?
Всё, на самом деле, довольно просто. В химии есть представление, что металлоорганикой можно считать только производные металлов, содержащие хотя бы одну связь металл-углерод.
Это хорошее определение, хотя и в нём нет стопроцентной ясности и определённости. Во-первых, что такое связь? Это мерзкий вопрос, вся двусмысленность которого продолжает раскрываться по мере того как современная химия узнаёт всё больше о взаимодействии атомов в молекулах. Есть у меня гадкое подозрение, что лет ещё через 50 химики могут вообще отказаться от понятия химической связи, настолько расплывчатым оно может стать к тому времени. В существенной мере это понятие можно считать реликтовым остатком старой химии 19 века, когда не было никакого представления об электронах и квантовой природе взаимодействий между атомами в молекулах, а считалось, что атомы связаны некой неведомой силой взаимного сродства, действующей в пределах отведённой Природой валентности (ещё одно понятие из глубокой древности, смысл которого в рамках современных представлений достаточно условен). В современных расчётах структуры молекул и их свойств, в принципе, можно уже сейчас спокойно и не вспоминать про какие-то химические связи и валентности. Но пока ещё мы к ним настолько привыкли, что нам кажется странным говорить о химии без этого. Но время идёт, люди привыкают в более абстрактным и менее укоренённым в прошлом понятиям, так что время придёт… К счастью, в ту пору прекрасную жить мне не придётся, а что вы будете тогда делать меня не очень волнует – разберётесь как-нибудь. Но даже сейчас и не забираясь со всякие тонкие и многоцентровые взаимодействия, спросим, а ионная связь тоже учитывается? Это ведь важно, но в первую очередь, для комплексов непереходных металлов. Слава всем Уполномоченным Богам, нас в этом курсе не волнуют производные непереходных металлов. А движуха там, в этой химии, уже сейчас знатная. Оказывается, что и производных у непереходных металлов, даже таких вроде бы примитивных, как щёлочноземельные металлы, целая пропасть, и структуры у них бывают – палладий позавидует! В принципе, среди переходных металлов есть третья группа, самая первая в d-блоке, группа скандия, которая очень близко смыкается с непереходными металлами. Немного ниже мы к этому вернёмся.
Во-вторых, а любая ли связь с углеродом есть признак органической химии. Это тоже старый спор: все ли соединения углерода суть органика, или, например, оксиды углерода, карбонаты, и тому подобные простые вещества не суть органика. На мой взгляд, это полная казуистика, схоластика, и канцелярия в одном флаконе. Нельзя разделить соединения углерода, особенно в современной химии. Старая химия ещй могла сказать, что, например, оксид углерода (II) ей не очень интересен, реакций де с ним мало, но современный химик умрёт от смеха от такого заявления. То же и с угольной кислотой. Разве не все органические вещества на Земле имели общего предка, молекулу CO2, как все люди единую праматерь, митохондриальную или обыкновенную Еву (и то, и другое – околонаучные метафоры)? Разве не считается совершенно официально получение мочевины Вёлером первой реакцией синтеза органического вещества в лаборатории? Ах, нет, ведь там не так – не просто первой органического, а первой органического из неорганических. Ах, тогда считали органическим только то, что имеет отношение к живым организмам, даже если ими было отвержено и извержено. И что, мы до сих пор сможем на полном серьёзе считать органическим веществом амид неорганичекой кислоты? К чёрту все это! Будем просто считать, что если есть какая-то внятная связь (такая, которую можно обозначить полноценными валентными черточками, количество которых в структуре должно соответствовать представлениям о законной валентности в рамках Периодического закона элементов, которую мы пока отменять не будем) между металлом и углеродом, то соединение можно считать металлоорганикой.
Итак, металлоорганикой будем считать координационные соединения металлов (нас интересуют переходные, но для общности возьмём все, включая металлоиды типа бора, кремния, и т.п.) имеющие хоть одну связь (ковалентную или координационную) металл-углерод.
Это определение не имеет статуса закона, но неявно разделяется большинством исследователей, имеющих дело с веществами такого типа. Например, именно так отбирают статьи для публикации в журналах со словом металлоорганика в названии (Organometallics, Journal of Organometallic Chemistry).
L-Лиганды
Разберемся поточнее, что стоит за этими обозначениями. Лиганд L-типа – это самый классический лиганд, молекула, в которой есть атом с неподеленной парой электронов. Это основание Льюиса, а в органической химии такие частицы называют еще нуклеофилами. Пока оставим очень тонкий вопрос, есть ли между этими понятиями сущностное различие, просто примем, что, если в нейтральной молекуле есть атом с неподеленной парой, то такая молекула может быть лигандом L-типа. Самое базовое представление о том, как образуются комплексы состоит в том, что один из партнеров предоставляет пустую орбиталь (т.е. является кислотой Льюиса), а второй – неподеленную пару (т.е. является основанием Льюиса). Тогда образуется химическая связь, которую часто называли и до сих пор иногда называют координационной. Поскольку нас интересуют комплексы металлов, то у нас всегда будет так, что металл предоставляет вакантную орбиталь, а лиганд L-типа пару.
Итак, L-лиганд должен иметь атом с неподеленной парой, то есть быть основанием Льюиса или нуклеофилом. Слово “нуклеофил” в этом контексте вызывает ассоциации с органической химией, а в ней, мы помним, нуклеофилы бывают сильные и слабые. В координационной химии такая простая модель не работает. Металлов много, а у каждого есть еще и по несколько валентных состояний, и способности металлов находить себе лиганды по вкусу гораздо шире. В общем, мы можем принять, что если у какой-то молекулы найдется атом с неподелённой парой, то найдется и металл, который этим не побрезгует, и охотно примет в свою координационную сферу. Да, многие лиганды этого класса и в органической химии назвались бы нуклеофилами: это и вода, и спирты, и эфиры, и аммиак, и амины, и фосфины, и т.п. Но вот оксид углерода органики редко используют как нуклеофил, а для переходных металлов, особенно поздних, это один из самых любимых лигандов. Азот – нуклеофил? Да-да, в курсе органической химии самые любопытные слышали что-то невнятное про обратимость диссоциации солей диазония, как будто бы арильный катион может обратно связать азот в диазоний, но это такая экзотика! Это же одна из лучших уходящих групп в органике! Уж уйдет так уйдет, назад не воротишь. А комплексов переходных металлов с азотом немало, настолько немало, что уже давно перестали они быть экзотикой. Да и почему нет, ведь азот (правильнее молекулу азота называть диазот, dinitrogen) по структуре почти неотличим от оксида углерода: они изоэлектронны и изоструктурны, да и вообще принадлежат к, наверное, самой восхитительной по многообразию и фундаментальной роли в химии и жизни группе двухатомных лигандов: нитрозоний, диазот, оксид углерода, цианид-ион, ацетиленид.
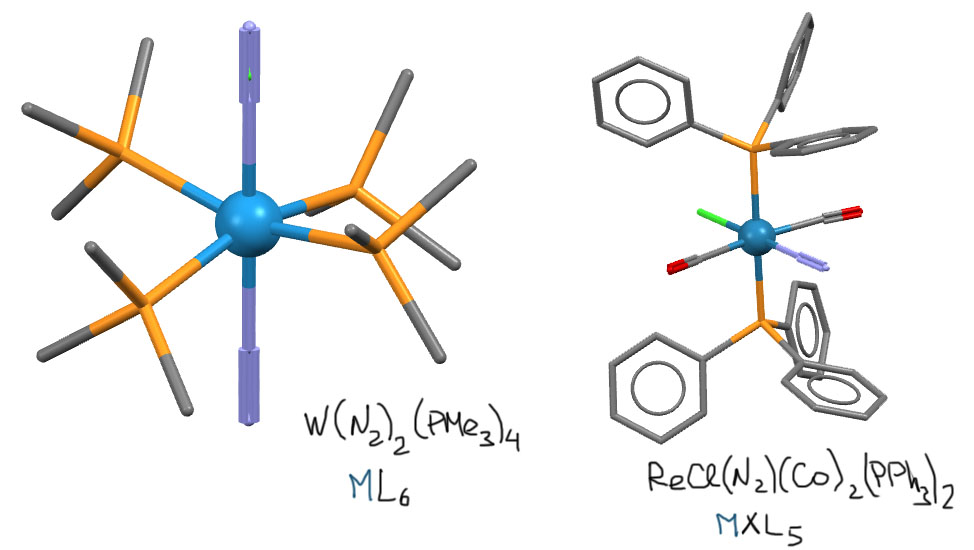
 Вот для примера пара реальных структур (атомы водорода не показаны) комплексов молекулярного азота, вольфрама (E.Carmona, J.M.Marin, M.L.Poveda, J.L.Atwood, R.D.Rogers, Polyhedron (1983), 2, 185) и рения (A.M.Kirillov, M.Haukka, M.F.C.G.da Silva, J.J.R.F.da Silva, A.J.L.Pombeiro, J.Organomet.Chem. (2006), 691, 4153). Второй комплекс включает оба изоэлектронных лиганда – карбонил и диазот, и очень хорошо видно, как они на самом деле похожи.
Вот для примера пара реальных структур (атомы водорода не показаны) комплексов молекулярного азота, вольфрама (E.Carmona, J.M.Marin, M.L.Poveda, J.L.Atwood, R.D.Rogers, Polyhedron (1983), 2, 185) и рения (A.M.Kirillov, M.Haukka, M.F.C.G.da Silva, J.J.R.F.da Silva, A.J.L.Pombeiro, J.Organomet.Chem. (2006), 691, 4153). Второй комплекс включает оба изоэлектронных лиганда – карбонил и диазот, и очень хорошо видно, как они на самом деле похожи.
X-лиганды
Лиганды X-типа не менее важны. Если образование координационной связи между металлом и L-лигандом выглядит однозначно как предоставление лигандом своей электронной пары, то связь металл – X-лиганд можно образовать двумя разными путями:
- как и обычная ковалентная связь она может рассматриваться как результат рекомбинации радикалов, по одному электрону от каждого;
- как координационная связь она может образоваться взаимодействием анионного лиганда с металлом, несущим формальный положительный заряд (пока не будем волноваться по поводу того, что такое “формальный заряд” – это одна из вещей в координационной химии, которая реально сводит с ума людей, привычных к четкой системе обозначения структуры, принятой в органической химии, где заряд всегда настоящий, а не формальный, – ведь мы не хотим раньше времени сходить с ума).
Слово “образовать” здесь не означает буквально “получить в реакции”, а показывает, как в принципе может образоваться такая связь, показывает ее сходство с тем, что мы в органической химии называем ковалентной связью.

X2-лиганды
Если L-лиганды все в первом приближении одинаковы – связаны с металлом координационной связью и не изменяют степень окисления, то с X-лигандами это не проходит. Как, например, устроен перманганат-ион, если посмотреть на него, как на координационное соединение, а как еще на него можно смотреть?
Тогда это тетраэдрический комплекс марганца с четырьмя одинаковыми лигандами с зарядом -1 и степенью окисления марганца +7. такой лиганд называется оксо, и весь комплекс будет тетраоксомарганец(+7) (или тетраоксоманганат(+7), но современная номенклатура предпочитает в основе названия координационного соединения название собственно металла без всяких суффиксов типа -оний или -ат, и это правильно, потому что заряд – сугубо формальная вещь, просто баланс электронов металла и лигандов, однозначно определяемый степенью окисления – указывать и степень окисления и заряд избыточно). То есть формально его придется рисовать с зарядом на марганце, хотя мы привыкли к структуре с зарядом на кислороде, но при этом имеем в виду, что реально заряд делокализован по всем четырем атомам кислорода точно так же, как в карбоксилате по двум атомам. В карбоксилате мы это изображаем резонансными структурами, и точно так же могли бы это сделать для перманганата.
Впрочем стоп. А как вообще рисовать связь с таким лигандом? В координационной химии это не праздный вопрос, хотя бы потому что природа связей в координационной химии более сложная, чем в химии неметаллов и p-элементов. Мы это еще увидим, когда перейдем к лигандам, связанным не через один, а через несколько атомов, и будем думать, сколько связей связывает металл и, например, циклопентадиенил-анион. Работает ли в координационной химии понятие порядка связи, и если работает, то нужно ли его отображать несколькими черточками и говорить о кратных, двойных и тройных связях? А как быть тогда с дробными порядками?
Еще яснее проблема вырисовывается, если сравнить перманганат-ион и структурно аналогичный пербромат-ион (даже называются почти одинаково, впрочем, это несистемные названия, берем немного экзотический пербромат вместо банального перхлората, чтобы оставаться в пределах одного периода). Оба тетраэдры, и… и сказать даже больше нечего, так это просто и очевидно. Даже длины связей практически одинаковы. Так и хочется назвать эти ионы не только изоструктурными, но и изоэлектронными. Но это было бы ошибкой. Бром – p-элемент, подчиняющийся правилу октета Льюиса, а следовательно такое его производное, как пербромат – пример так называемой гипервалентности. Иными словами, когда мы в пербромате или перхлорате (или сульфате, которому можно противопоставить манганат или хромат анионы) рисуем двойные черточки, это просто дань традиции, не имеющая отношения к реальной электронной структуре. У брома (хлора, серы) с валентными sp-оболочками просто нет валентных возможностей для структуры с двойными связями. Но у марганца, хрома и других переходных металлов такие возможности как раз есть. И нет никаких проблем в настоящей двойной связи металл-кислород, которую можно честно изобразить двумя черточками. Это очень хорошо, потому что мы, например, тогда видим почти полную (почти в данном случае означает, что элемент 1 и 3 рядов используют одни и те же электроны, но с разных валентных оболочек) изоструктурность и изоэлектронность перманганата (тетраоксомарганец(+7)) и тетраоксида осмия (тетраоксоосмий(+8)), и больше не удивляемся тому общеизвестному факту, что эти два реагента совершенно одинаково реагируют с двойной связью в олефинах. Еще раз обращаю внимание на то, что когда мы рисуем заряд, особенно минус, на металле, это просто для экономии места (нужно было бы заключить комплекс в квадратные скобки и поставить минус за ними), и ни в коем случа не значит, ни в этом комплексе, ни в других, что этот минус сидит на металле. Где на самом деле сидит этот минус, это всегда отдельный вопрос, и далеко не очевидный.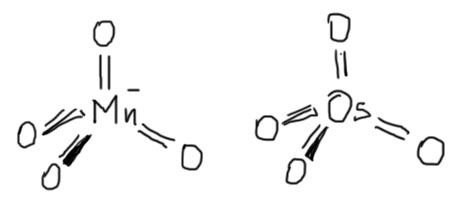
Каждой черточке соответствует вполне конкретное взаимодействие – σ-связь, образованная dσ-орбиталью металла и sp2-гибридной орбиталью кислорода, и π-связь, образованная dπ-орбиталью металла и p-орбиталью кислорода. На резонный вопрос, откуда такая чертова пропасть этих dσ– и dπ-орбиталей у одного металла, заметим просто, что если бы у нас был комплекс с только одним таким лигандом, такое описание было бы совсем адекватным, а если таких лигандов много, например, четыре, то имеющиеся атомные орбитали такой симметрии просто образуют многоцентровые МО комплекса. Поскольку мы договорились не вдаваться в подробности квантово-химического описания структуры, придется в таком сыром виде это и оставить. С нашей точки зрения важно то, что лиганд, подобный кислороду (оксо-лиганд):
- изменяет степень окисления металла на 2 единицы в плюс;
- в ионном счете электронов дает 4 электрона, в ковалентном – два
- и наконец, занимает одно – не два! – координационное место в координационной сфере металла, и так и должен учитываться в координационном числе, например, к.ч. марганца в перманганате – четыре, а не восемь;
Если все это принять во внимание, то получится, что такой лиганд почти по всем параметрам эквивалентен двум X-лигандам. И если бы не было слова “почти”, можно было бы вообще не выделять такие лиганды в особую группу. Но с точки зрения места в координационной сфере это не так – это именно один лиганд, а не два. Поэтому этот тип и называют X2. Иными словами, структурный тип перманганата не MX8, что подразумевало бы очень редкое к.ч. 8, свойственное, и то редко, элементам 3 ряда переходных металлов, а M(X2)4, и к.ч. 4, и это вездесущий тетраэдр. У тетраоксида осмия и перманганата структурный тип одинаков.
Лиганды X2-типа встречаются гораздо реже просто X-лигандов. Самый распространенный из них как раз оксо-лиганд, часто встречающийся в оксо-комплексах высоких степеней окисления. И это не только хорошо известные молекулярные оксиды типа тетраоксида осмия, кислородные кислоты металлов 5-8 групп и их соли (ванадаты, хроматы, молибдаты, вольфраматы, манганаты, перманганаты, перренаты, и т.п.), производные кислородных катионов типа титанила, ванадила, хромила и т.п., но и более сложные комплексы с кучей других лигандов. Такие комплексы часто встречаются во всевозможных, каталитических и некаталитических реакциях окисления.
Такой же лиганд есть с серой (тио-лиганд), но он встречается намного реже, потому что сера предпочитает мостиковые формы связывания. Как и в обычной органической химии, двойная связь с другими элементами для серы менее характерна, чем для кислорода.
А вот у азота таких лигандов очень много потому что еще одна валентность несет заместитель, обеспечивая огромную вариантность. Такие лиганды называют -имидо лигандами. Это не самое лучшее название, так как оно вызывает двусмысленное толкование – имидом можно считать не только двухвалентный остаток на азоте, но и производное имина. Проблема есть, но в химии вообще много двусмысленностей, закрепленных традицией. Мы встретимся с ними как минимум один раз – в метатезисе. Для тренировки разбора структуры приведу пару комплексов с X2-лигандами: 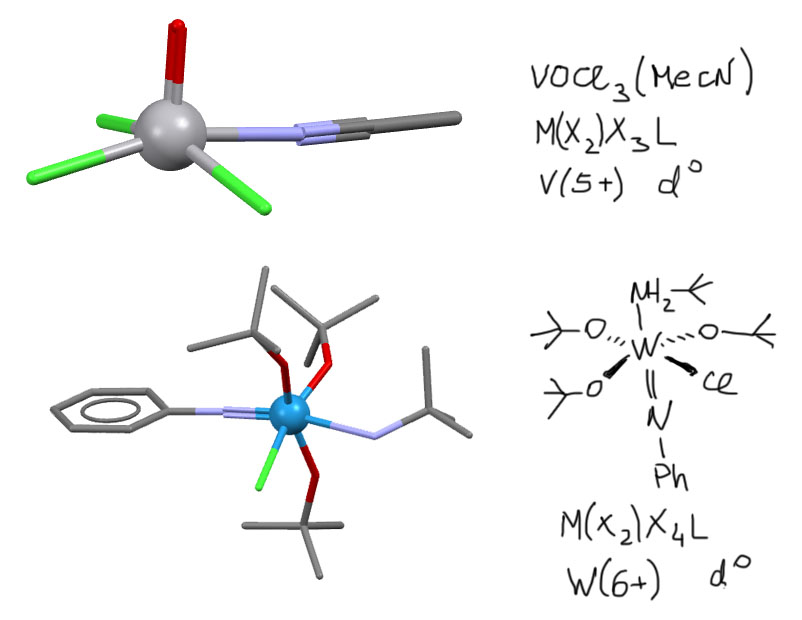 (комплекс ванадия: J.C.Daran, Y.Jeannin, G.Constant, R.Morancho, Acta Crystallogr.,Sect.B:Struct.Crystallogr.Cryst.Chem. (1975), 31, 1833; вольфрама: A.J.Nielson, J.M.Waters, Polyhedron (1982), 1, 561).
(комплекс ванадия: J.C.Daran, Y.Jeannin, G.Constant, R.Morancho, Acta Crystallogr.,Sect.B:Struct.Crystallogr.Cryst.Chem. (1975), 31, 1833; вольфрама: A.J.Nielson, J.M.Waters, Polyhedron (1982), 1, 561).
А что у углерода? Вот с углеродом все очень сложно. Продолжая ряд кислород-азот-углерод, можно понять, что лиганд подобного типа будет нести два заместителя. Углерод с двумя заместителями – это карбен. Карбены – очень популярные и очень важные лиганды для переходных металлов. И они бывают лигандами как X2-типа, так и L-типа. Разберемся с этой непростой проблемой позже.
X3-лиганды
Если есть X2-лиганды, связанные с металлом двойной связью, то возможно есть и троесвязанные лиганды. Довольно очевидно, что искать их надо среди производных азота. Режем молекулярный азот пополам и приделываем к металлу. Такой лиганд ожидаемо называется нитридом. Он учитывается как один X3-лиганд,
- дает вклад +3 в степень окисления металла;
- учитывается как 6 электронов в ионном способе подсчета электронов, и 3 электрона – в ковалентном;
- занимает одно место в координационной сфере металла
Вот пара примеров нитридных комплексов. Первый забавен тем, что на один атом металла, молибдена, в нем 13 атомов азота и больше ничего, если не считать противоиона (K.Dehnicke, J.Schmitte, D.Fenske, Z.Naturforsch.,B:Chem.Sci. (1980), 35, 1070). Второй комплекс металла другой группы и другого состава, тем не менее, принадлежит к тому же структурному типу (P.A.Shapley, Z.-Y.Own, J.C.Huffman, Organometallics (1986), 5, 1269)
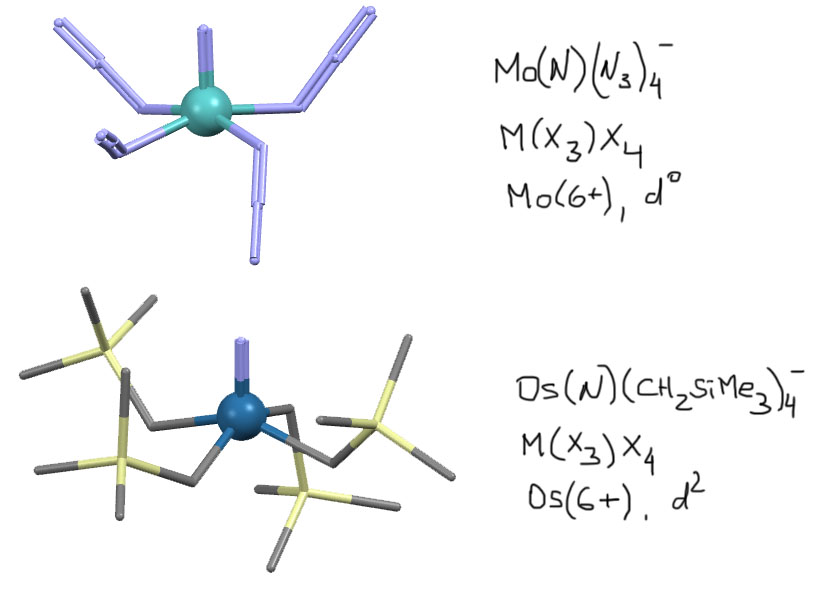 Кроме нитрида к тому же X3-типу относятся углеродные лиганды – карбины. Карбин – это половинка ацетилена, и аналогия с нитридом вполне очевидна. Отложим обсуждение карбиновых комплексов до реакции метатезиса алкинов, где они играют ключевую роль.
Кроме нитрида к тому же X3-типу относятся углеродные лиганды – карбины. Карбин – это половинка ацетилена, и аналогия с нитридом вполне очевидна. Отложим обсуждение карбиновых комплексов до реакции метатезиса алкинов, где они играют ключевую роль.
X3 лигандов становится все больше
До относительно недавнего времени X3-лиганды были ограничены только нитридом и карбинами. Но с 1995 года – это такой странный год в химии переходных металлов, когда все исследователи как будто с цепи сорвались. Очень похоже на взлёт очень тяжелого самолета, вид сбоку – сначала махина медленно катится по полосе как будто даже страшно медленно и без видимого ускорения, и кажется, что она вот так дальше и поедет до цели, снося дома, давя пешеходов, машины и проделывая просеки в лесах, но вдруг, фигак – и свечкой вверх, и сразу такой лёгонькой птичкой в небо.
Вот мы ещё увидим, какие важные вещи появились в этот год и как весело пошло дело дальше. И вот среди новых реакций и методов, еще и лиганды новые, и не только очень сложные, но и в некотором смысле самые простые, проще некуда, одноатомные. Например, фосфор на тройной связи, аналог нитрида. К сожалению основатели химии фосфора не предугадали, насколько сложной и обильной будет химия этого элемента и поскупились на корни слов. Вот в химии азота есть греческий корень азот, азо, и латинский нитро. И мы элементарно разводим одновалентные X-лиганды, амиды, и трехвалентные X3-лиганд, нитрид. А с фосфором понадеялись на то, что такого лиганда нет и не будет никогда, и фосфиды останутся фосфидами – аналогами амидных лигандов. А как тогда назвать аналоги нитридного лиганда? В способность фосфора и его более тяжелых аналогов образовывать настоящие тройные связи долго не верилось, фосфор – более крупный элемент, чем азот и углерод, свяи длиннее, и нормально организовать перекрывание, соответствующее короткой тройной связи казалось невозможным.
Всё изменилось, когда Ричард Шрок, один из главных основоположников метатезиса, стал копать в координационной химии, пытаясь развить свой успех с карбенами и карбинами Шрока (об этом потом). Поскольку закономерности стабилизации таких лигандов стали понятны, эти знания стали пробовать применить к аналогам с соседними элементами. На тройную связь может теоретически претендовать любой элемент группы углерода и группы азота – нужно было поискать аналоги карбинов и нитрида. И это сработало, прежде всего на фосфоре. В 1995 году и сам Шрок и его ученик Кристофер Каминс, который дальше в основном и потащит это направление, опубликовали в двух статьях друг за другом (обратите внимание, как Шрок благородно пропускает ученика вперёд, и не пишется в соавторы, хотя эта работа целиком сделана на его идеях) два первых комплекса с фосфором на тройной связи. Для этого использовали опыт работы с карбинами, то, что лучше всего подходят тяжелые элементы 6-й группы, особенно вольфрам. Эти элементы, с одной стороны, проявляют свойства ранних переходных металлов и степень окисления 6+, но с другой стороны, весьма электроотрицательны, а это не позволяет лигандам распоясаться и отнять у металла лишнюю электронную плотность, образовав вместо тройной связи, например, двойную с разделением зарядов и высокой ионностью.
Вот они оба;
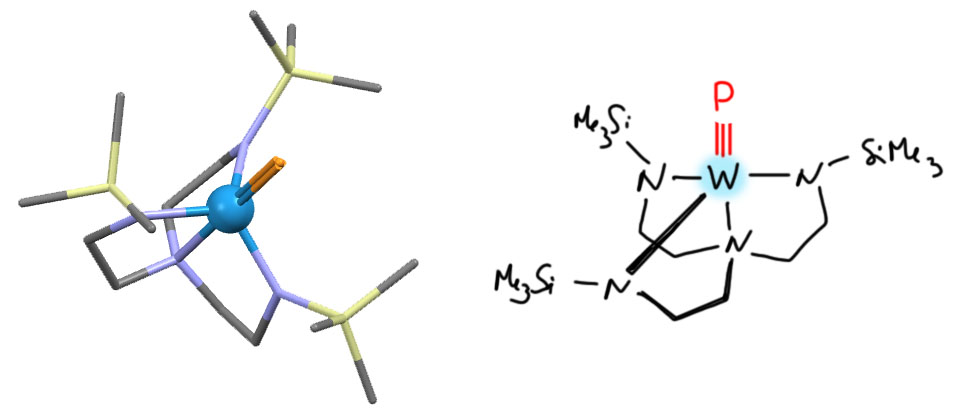
N.C.Zanetti, R.R.Schrock, W.M.Davis, Angew.Chem.,Int.Ed. 1995, 34, 2044, код структуры ZEQXEY
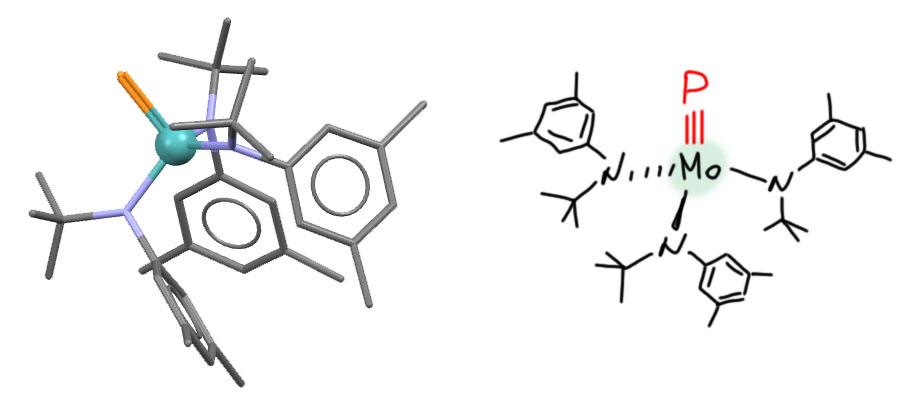
C.E.Laplaza, W.M.Davis, C.C.Cummins, Angew.Chem.,Int.Ed. 1995, 34, 2042, код структуры ZEQWUN
После этого дело пошло. Комплексов с X3-фосфидным лигандом с 1995 года наделали немало, и появилось целое направление в химии, изучающее лиганды такого типа в группах углерода и азота. Не у всех элементов такие лиганды найдены, и чем ниже элемент, тем шанс ниже по очевидной причине – атомы действительно становятся очень большими, а связи длинными, и обеспечить эффективное перекрывание становится непросто. Подробно мы туда не полезем хотя бы потому, что в органической химии это пока что не очень применяется, но можно поспорить, что применение таким комплексам найдут, и очень скоро. Химия у них весьма впечатляющая, например, фосфидные комплексы умеют разбирать молекулу белого фосфора на части, образуя целую серию комплексов с очень интересными структурами.
А здесь приведу еще комплекс с мышьяком на тройной связи, описанный совсем недавно тем же Камминсом. Противоион не показан, но приблудился диоксан из растворителя.
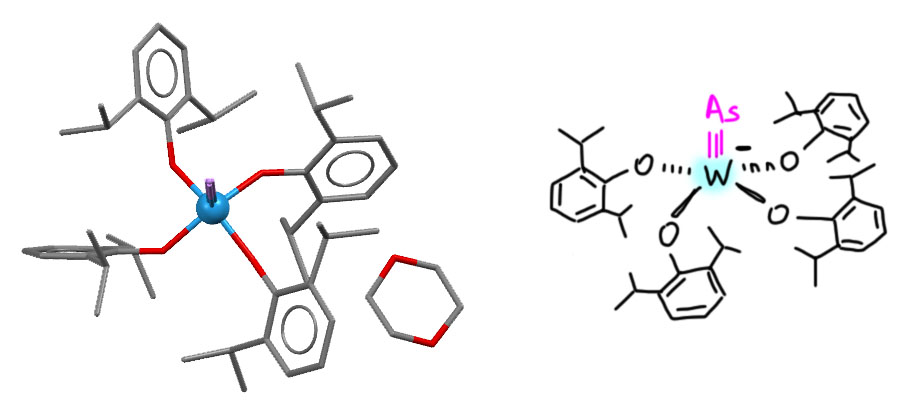
M.Joost, W.J.Transue, C.C.Cummins, Chem.Commun. 2017, 53, 10731, код структуры YEHFEZ
В связи с лигандами X-типа возникает такой же вопрос, как и по отношению к обычным ковалентным связям: в какую сторону смещена электронная плотность. В химии для грубой оценки смещения используют шкалу электроотрицательностей. И здесь нас могут ожидать сюрпризы. Довольно широко распространено поверие, что любой неметалл более электроотрицателен, чем любой металл. Это довольно далеко от реального положения вещей. Если посмотреть на положение элементов по шкале электроотрицательностей, то увидим, что самый важный элемент для органической химии, углерод, действительно более электротрицателен чем любые металлы за исключением золота, хотя и там разница минимальна. Но вот уже водород вполне проигрывает целой группе благородных металлов, и практически не отличается от не менее большой группы других, в основном поздних переходных металлов. Столь же или даже еще менее электроотрицательны элементы подгруппы фосфора, а кремний и бор уступают еще больше. Если же мы еще и подумаем о том, что электроотрицательность – скорее качественная чем количественная величина, и что в реальных соединениях на поляризацию связей влияет не только природа элементов, но и электронные эффекты заместителей или лигандов, то окажемся в непростой ситуации. К счастью, это не так важно, по крайней мере, для самой простой и очень важной вещи – определения степени окисления элемента, а следовательно и его электронной конфигурации.
Для определения степени окисления элементов в координационных соединениях и комплексах принимают, что металл, переходный или непереходный, всегда менее электроотрицателен, чем неметалл.

Определяем степень окисления металла и электронную конфигурацию.
Теперь мы легко определим степень окисления металла. Иногда говорят еще так – формальную степень окисления. Но неформальной степени окисления не существует, поэтому это добавление избыточно. Степень окисления – формальная величина, получающаяся в предположении, что все ковалентные связи поляризованы так что
- связь неметалл-неметалл поляризована в сторону более электроотрицательного элемента
- связь металл-неметалл поляризована в сторону неметалла
- связь металл-металл встречается относительно редко, но если встретится, то поляризована в сторону более электроотрицательного
Поскольку нас интересуют не любые молекулы, а только комплексы металлов, то все становится совсем просто.
- Берем комплекс и определяем типы лигандов, записываем обобщенную формулу комплекса – структурный тип.
- Лиганды L-типа не вносят никакого вклада в степень окисления
- Лиганды X-типа каждый добавляют по +1 в степень окисления
Правило 3 не абсолютно, но должно учитывать, с каким элементом связан металл, но для настоящих неметаллов от водорода и выше (точнее, правее) оно работает безусловно и всегда.
Попутно определим электронную конфигурацию. Это еще проще – берем номер группы и вычитаем из нее степень окисления.
Попробуем:

Проще не придумать. Впрочем, довольно часто бывает так, что комплекс имеет заряд. Это очень тонкое место, потому что просто наличие заряда на комплексной частице ничего не говорит о том, где этот заряд сидит – на металле или где-то на лигандах. Пока лиганды простые, кажется, что все просто и нет смысла усложнять. Но лиганды бывают не самые простые, и история с зарядами комплексов – одно из тех мест, где запутаться можно легко и быстро, и реально путаются даже довольно искушенные люди. Ограничимся пока теми случаями, когда с лигандами все ясно, а именно лиганды такие же, с которыми мы встречаемся в нейтральных комплексах.

И еще раз обращаю внимание на то, что степень окисления и намертво связанное с ней число электронов – понятия формальные, и именно так к ним и следует относиться, то есть очень серьезно. К формальным вещам вообще нужно относиться серьезно. Бесчисленное количество преступлений и даже ошибок было совершено и еще будет совершено, оттого что люди несерьезно относятся к формальным вещам. В реальном комплексе, например, в том же тетракарбонилкобальтат-анионе [Co(CO)4]– отрицательный заряд не будет сидеть на атоме металла, а будет делокализован на сильно акцепторных лигандах и в основном отъедет на атомы кислорода. Но способ рисования формул, принятый в координационной химии, в явном виде это никак не показывает, даже не имеет общепринятых способов изображения такой делокализации, в отличие от старой доброй органической химии. Мы всегда рисуем формальную формулу, извиняюсь за тавтологию, и присваиваем атомам формальные заряды, формальные степени окисления, формальные электронные конфигурации. Это было бы довольно бессмысленным упражнением, если бы не существовало общепринятой конвенции о том, как это делать всегда одинаковым образом. Любой химик всегда нарисует формулу комплекса, расставит заряды и степени окисления, посчитает d-электроны одинаково (бывают хитро устроенные комплексы, когда это непросто или даже невозможно, но их относительно мало, и с каждым из них можно разобраться отдельно). Это и делает формальное упражнение весьма полезным инструментом, пользуясь которым легко классифицировать реакции, находить стехиометрические отношения реагентов, понимать роль катализаторов, и т.п. А в тех случаях, когда нам нужно точнее описать структуру, определить реальное распределение зарядов, и тому подобные существенные вещи, мы это сможем сделать, зная как устроены связи в координационных соединениях, и подключив аппарат квантовой химии.