Олефины и ацетилены
Соединения с кратными углерод-углеродными связями являются одними из важнейших субстратов для реакций с участием комплексов переходных металлов. Количество таких реакций огромно и регулярно увеличивается. Изучение таких процессов – один из самых живых разделов органической химии. Среди реакций олефинов и ацетиленов очень много процессов промышленного значения, и не только в тонком органическом синтезе современных лекарственных препаратов и новых материалов, но и в крупнотоннажном синтезе полупродуктов, а также в полимеризации – в современной промышленности полимеров производство полимеров молекул с двойной связью (полиолефинов) с использованием комплексов переходных металлов в качестве катализаторов полимеризации. Как и в смежных областях химии комплексов переходных металлов в органическом синтезе, в последние 20 лет наблюдается бурный прогресс, связанный с качественным скачком в понимании закономерностей реакционной способности координационных соединений и рационального дизайна лигандов и реагентов новых типов.
Реакционная способность олефинов и ацетиленов после вхождения в координационную сферу металла определяется тонким балансом двух взаимодействий – прямой координационной связи и эффекта обратного донорного взаимодействия, отвечающие за противоположные направления смещения электронной плотности – от лиганда на металл и от металла на лиганд. Сначала посмотрим на два крайних случая, когда преобладает одно из взаимодействий. Преобладание обратного донорного эффекта (back-donation) сообщает координированному олефину свойства нуклеофила, а каждый из двух атомов углерода связи становится похож на карбанион.
Ещё раз повторим то, что мы уже изучили. Вхождение олефина или ацетилена в координационную сферу переходного металла приводит к изменению свойств и реакционной способности кратной связи. Результирующее изменение определяется балансом двух противонаправленных эффектов. Вход олефинов и ацетиленов в координационную сферу металла почти всегда осуществляется через η2-комплексы. Баланс двух взаимодействий - обычной координационной связи, обеспечивающей смещение электронной плотности с π-связи на металл, и обратного донорного эффекта (back-donation), обеспечивающего смещение плотности в обратную сторону - определяет реакционную способность координированного ненасыщенного соединения.
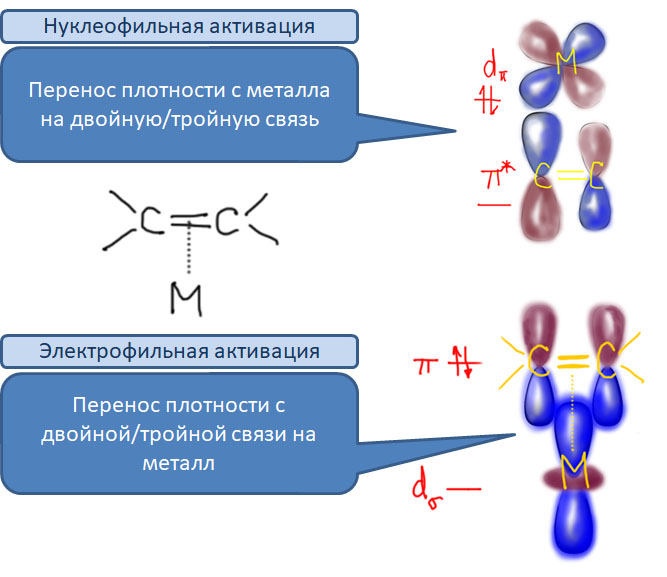
Преобладание back-donation делает кратную связь более богатой электронной плотностью, нуклеофильной, способной к реакциям с электрофилами. Этот эффект специфичен именно для переходных металлов.
Преобладание обычного координационного взаимодействия, в котором металл является кислотой Льюиса, напротив, уменьшает плотность на кратной связи, делает ее электрофильной, способной к реакциям с нуклеофилами. Этот эффект может наблюдаться не только с переходными металлами, но вполне типичен и для непереходных металлов.
В комплексах металлов с олефинами или ацетиленами с преобладанием back-donation в связывании происходит перенос электронной плотности с металла в сторону двойной или тройной связи, что сопровождается изменением гибридизации связанных с металом атомов углерода, и фактическим изменением степени окисления металла. Комплексы в этом случае получают структуру металлациклов - металлациклопропанов - в этом случае лучше уже говорить не о связи металла с кратной связью целиком, а с двумя отдельными ковалентными связями с атомами углерода кратной связи. Хотя так и не принято говорить, но кратная связь в этих случаях фактически превращается в бидентатный лиганд, а фрагмент металл-кратная связь выглядит как трёхчленный хелатный цикл - металлацикл.
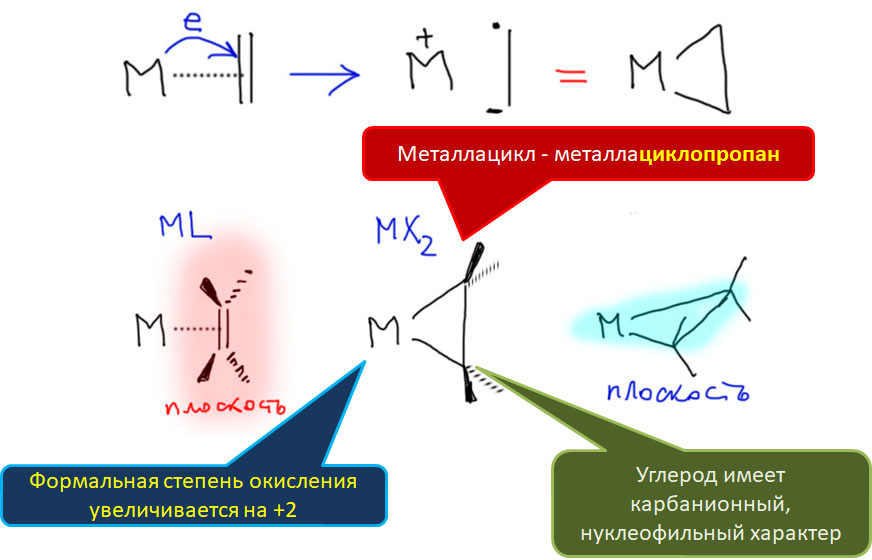
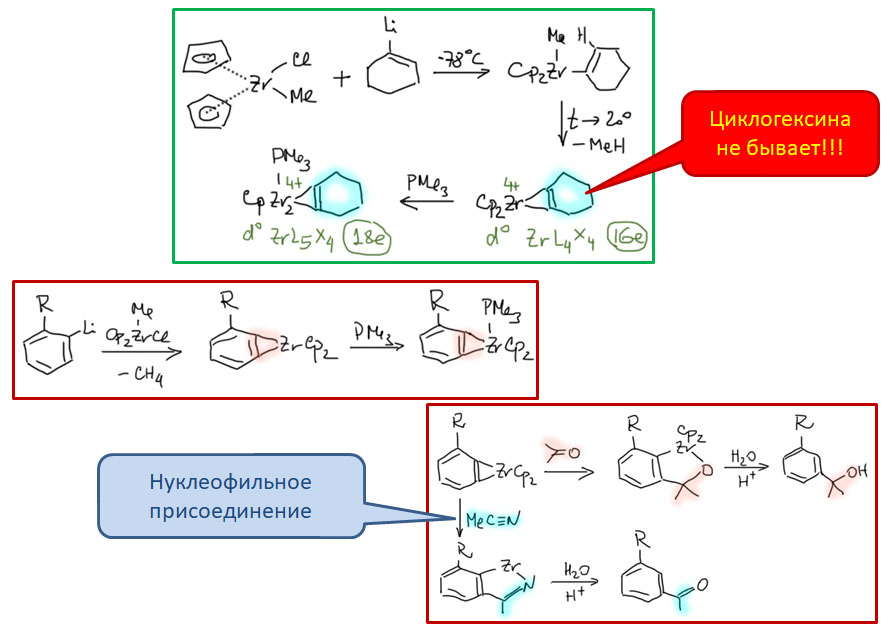
Особенно распространены такие комплексы у ранних переходных металлов, в особенности металлов 4 группы - титана, циркония, гафния. Комплексы этих металлов в исходном состоянии со степенью окисления +2 и конфигурацией d2 с двойной или тройной связью реально имеют структуру, соответствующую структурному типу MX2, а не ML, степенью окисления +4 и конфигурацией d0. Связи металл-углерод в таких комплексах приобретают высокую степень ионности, а углероды - выраженный карбанионный характер, как в типичных металлоорганических соединениях электроположительных непереходных металлов типа алюминия или даже магния.
Более удобный способ получения металлациклопропанов и -пропенов состоит в реакции переметаллирования алкильных комплексов M(4+) c литийорганическими соединениями, за которой обычно следует элиминирование алкана. Так получают, в том числе и комплексы с алкинами, которые не являются устойчивыми соединениями, например, с циклоалкинами с небольшим размером цикла и с дегидробензолами.
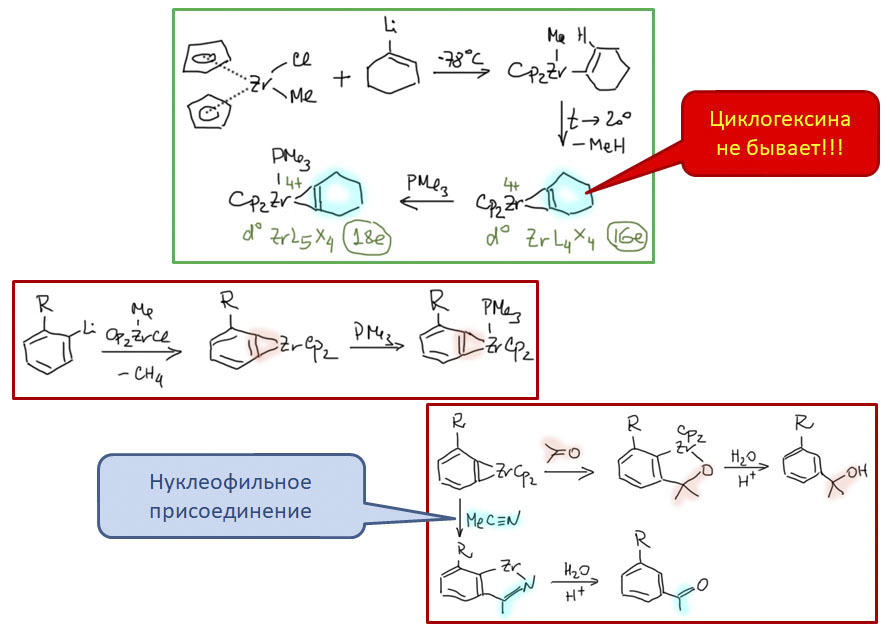
Углероды кратной связи приобретают отчетливый карбанионный характер, и превосходно реагируют с электрофилами разного типа в реакциях, очень похожих на реакции магнийорганических соединений. Комплексы дегидробензолов с орто-заместителем при этом направляют электрофил на дальний атом углерода по стерическим причинам.
Синтез циклопропанов по Кулинковичу - - изумительный пример превращения с участием металлациклопропанов. Обычная реакция сложных эфиров с двумя молями реактива Гриньяра, дающая третичные спирты, приводит к совершенно другому, и на первый взгляд, невероятному продукту, если в реакционной смеси присутствует изопропилат титана. Механизм реакции показывает, что мы имеем дело с образованием титанациклопропана, который далее обоими углеродами нуклеофильно атакует карбонил сложного эфира точно так же, как это делал гриньяр.
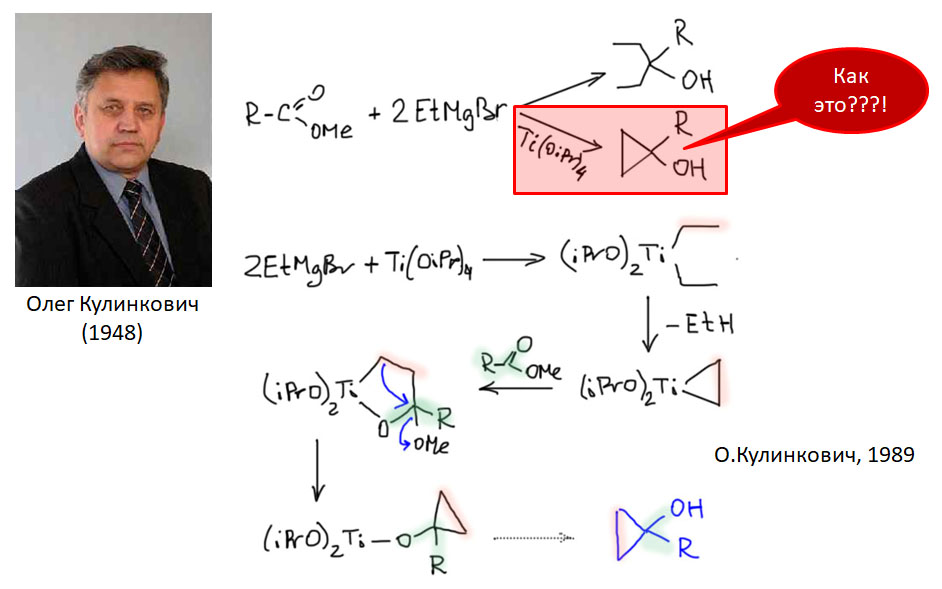
Реакция исключительно проста и эффективна, став в синтезе лучшим методом синтеза третичных циклопропанолов, а это очень важный класс производных циклопропана, участвующий во многих сложных синтезах. Развитие реакции Кулинковича привело к более общему методу (реакция Кулинковича-де Мейере) синтеза циклопропанов и не только циклопропанов.

Электрофильная активация кратных связей не менее важна и лежит в основе многих важных процессов, так как такая активация приводит к тому, что к активированным кратным связям присоединяются даже слабые нуклеофилы. Дальнейшее развитие сюжета зависит от металла и от способа выхода сигма-связанного органического лиганда, образовавшегося в результате такого присоединения, из координационной сферы металла.
Реакция внешнего (то есть, не находящегося в момент реакции в координационной сфере металла) нуклеофила с активированной кратной связью в координационной сфере металла, который в этом случае проявляет свойства π-кислоты (кислоты Льюиса, связанной в π-комплекс с кратной связью, хотя мы же помним, какая здесь путаница, потому что связь кратной связи с металлом это сигма-связь, но так уж это по традиции называют). В результате реакции образуется новый комплекс с σ-связью металл - алкил (или винил), а нуклеофил связывается со вторым атомом углерода. Стереохимия таких реакций - всегда анти (транс).
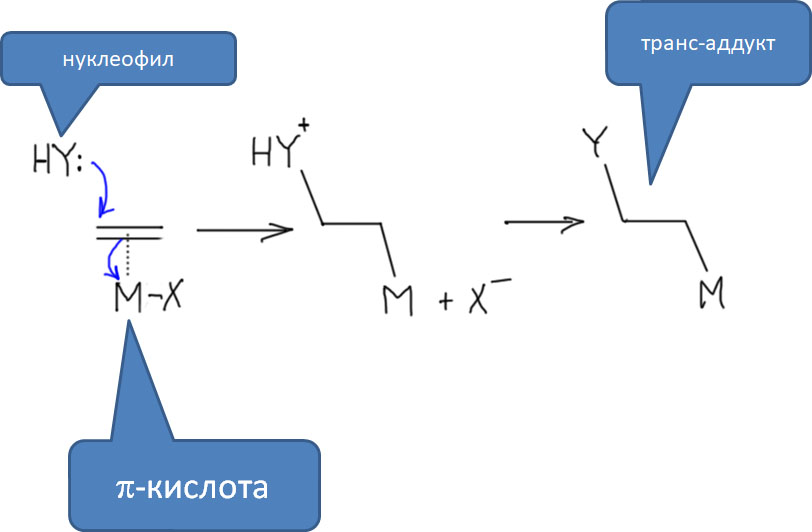
Реакции этого типа называют чудным словом - нуклеометаллированием, имея в виду, что присоединяется нуклеофил и металл. В конкретных случаях применяют более точные термины, явно называя и нуклеофил, и металл.
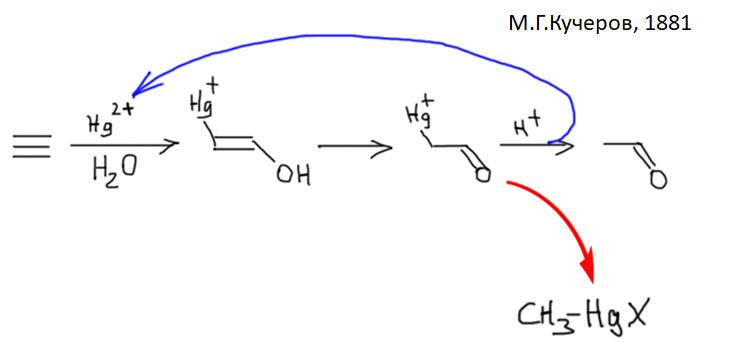
У реакции нуклеометаллирования есть очевидные прототипы в обычной органической химии. Самый близкий - реакция Кучерова, открытая в 1881 году. Роль этой реакции, которую обнаружил и описал химик-любитель Михаил Кучеров, в дальнейшем развитии органической химии трудно переоценить - фактически это первый пример органической реакции, катализируемой комплексом металла. Хотя ртуть - это непереходный металл (но d-элемент), ее уникальная способность образовывать очень прочные связи с углеродом делает отчасти похожей на переходные металлы.
В результате получился каталитический метод синтеза очень важного соединения, ацетальдегида, из ацетилена, который в начале органического синтеза рассматривали как очень привлекательное сырье, потому что он дает возможность получать ценные органические молекулы фактически из угля и воды. В дальнейшем развитии химической промышленности, когда объемы производства перешли от сотен килограммов и тонн к тысячам тонн, от ацетилена практически отказались из-за огромной энергоемкости его синтеза и опасности работы.
Но в начале 20 века эта реакция стала активно использоваться в промышленном производстве ацетальдегида. Результатом стала, вероятно, самая трагическая катастрофа в истории химической промышленности. В реакции Кучерова побочно в небольших количествах образуется производное метилртути, которое стало попадать в сточные воды и накапливаться в окружающей среде. В японском местечке Минамата, рядом с которым находился химический завод компании Тиссо (Chisso), сточные воды отравили залив, что привело к массовому отравлению местных жителей, в первую очередь, детей. Более 1700 человек умерло мучительной смертью, другие остались искалеченными.
Ртуть - плохой металл для промышленности.
В 1950-х Герберт Браун описал реакцию сольвомеркурирования, аналогичную реакции Кучерова, но использующую олефины, а не ацетилены. Сольвомеркурирование дает возможность получать продукты присоединения нуклеофильных растворителей: воды, спиртов, карбоновых кислот, и даже аминов сильно разветвленной структуры, а это очень непросто сделать другими способами. Поэтому эта реакция нашла широкое применение в синтезе, даже несмотря на ее стехиометрический характер и необходимость использовать огромные количества солей ртути.
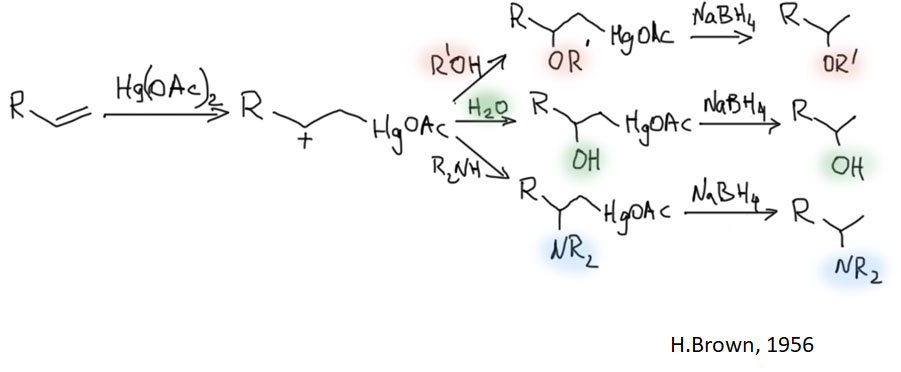
В органической химии эти реакции расматривали как электрофильную атаку на кратную связь с последующей "подсадкой" нуклеофила на образующийся карбокатион или циклический (меркуриниевый) ион. В химии переходных металлов принятая последовательность событий та же самая, но роль металла сводится к активации кратной связи к атаке нуклеофила, а само присоединение нуклеофила становится центральным событием реакции нуклеометаллирования - в этих случаях мы назовём это гидроксимеркурированием, алкоксимеркурированием, аминомеркурированием.
В современных реакциях нуклеометаллирования чаще всего используются в качестве π-кислот комплексы золота и платины. “Золотая” реакция Кучерова, например, катализируется многими комплексами золота, может иметь огромные величины TON и TOF.
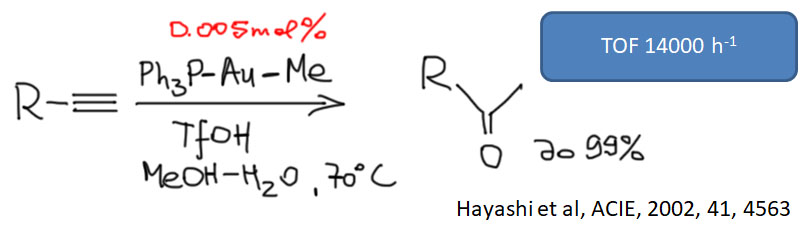
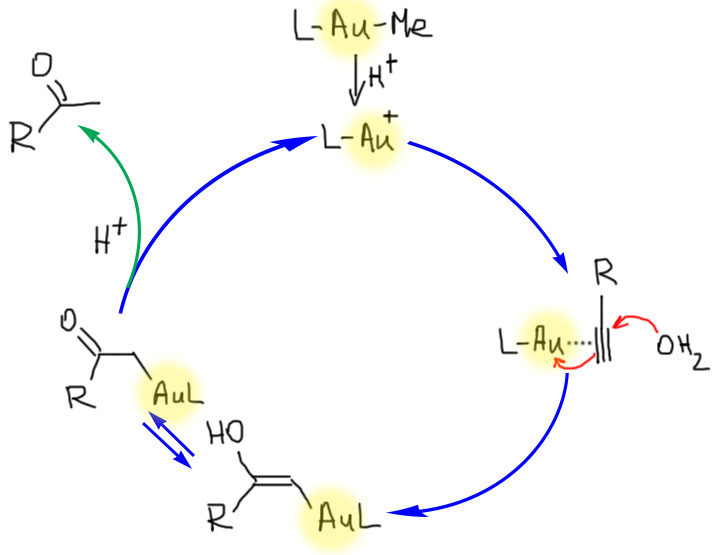
Комплексы золота, в основном относящиеся к структурному типу AuXL, обладают непревзойденными качествами в таких реакциях. X-лиганд подбирают так, чтобы в условиях реакции он диссоциировал с образованием каталитически активного положительно-заряженного комплекса, координирующегося по кратной связи, активируя ее для нуклеофильного присоединения, например, воды. Дальше происходит ровно то же самое, что в реакции Кучерова. Расщепление (протолиз) связи углерод-золото регенерирует каталитически активный комплекс золота. Реакция происходит в присутствии сильных кислот.
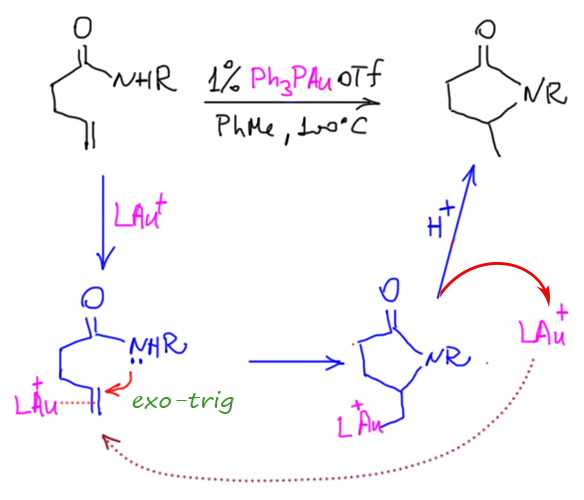
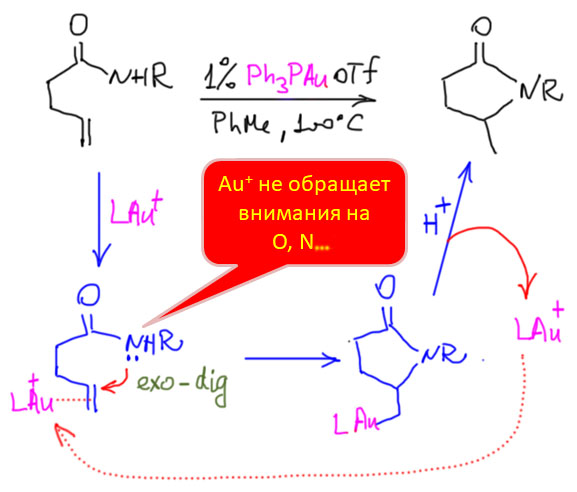
Совершенно потрясающим свойством комплексов золота(1+) является то, что, действуя как кислота Льюиса, они предпочитают двойные и тройные углерод-углеродные связи и совсем не замечают нуклеофильных центров, на которые тут же запали бы почти все другие кислоты Льюиса - атомы азота или кислорода. Именно поэтому их прозвали карбофильными - то есть любящими углерод.
Особенно легко такие реакции проходят во внутримолекулярном варианте - в виде циклизаций нуклеофильных центров на кратные связи. По очень популярной классификации Болдуина циклизацию обозначают как экзо (если один из углеродов связи не включается в цикл), или эндо (если включаются оба). И если циклизация идёт на двойную связь - то называется триг (от тригональный, то есть sp2-углерод).
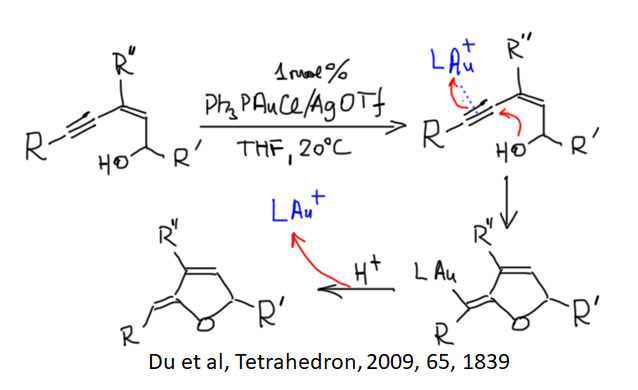
А вот пример циклизации на ацетиленовый фрагмент с присоединением гидроксила. Это тоже экзо-, но диг-циклизация (от дигональный - то есть буквально двуугольный, то есть прямой, то есть просто sp-углерод). Циклизация в данном случае принадлежит к типу экзо-диг. Исходный комплекс LAuCl активируют трифлатом серебра - замещение лиганда хлорида на трифлат, который не держится на металле и освобождает свободное координационное место, как в предыдущем примере, но с использованием более доступного исходного комплекса.
По счастливому совпадению, когда возникли огромные проблемы с получением ацетальдегида по Кучерову, две немецкие компании, Вакер и Хёхст разработали новый метод синтеза ацетальдегида из гораздо более дешёвого и безопасного этилена.
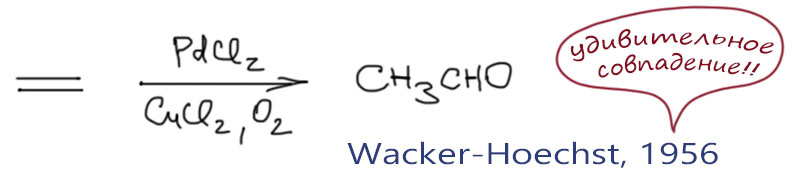
Хлорид палладия в сильнокислой среде превращает этилен в ацетальдегид по механизму нуклеометаллирования - комплекс этилена с хлоридом палладия, очень похожий на знаменитую соль Цайзе, присоединяет воду. Но образующийся сигма-комплекс палладия неустойчив, так как очень легко элиминирует гидридный комплекс палладия. Немного позже мы разберёмся в этой очень важной элементарной реакции, очень часто встречающейся в каталитических процессах с участием не только палладия, но и почти всех других переходных металлов.
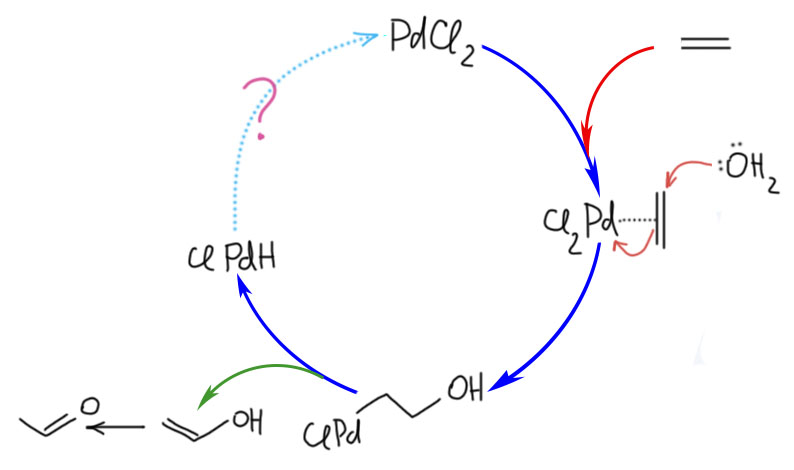
Но вот проблема - а как вернуть палладий на старт процесса, как превратить гидридный комплекс в просто хлорид. Пока это не сделано, процесс является некаталитическим, и на превращения моля этилена понадобится моль хлорида палладия. Ясно, что у такой реакции перспектив не было бы. Нужно как-то замкнуть процесс в цикл.
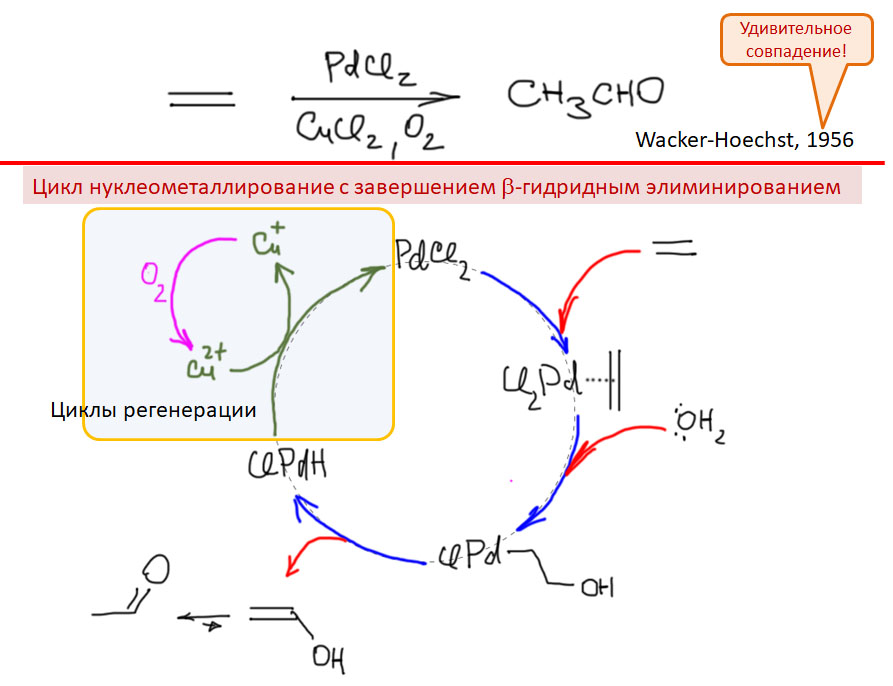
Превращение гидридного комплекса палладия в хлорид палладия - это окисление. Хотя в обоих комплексах Pd(2+), окисляем мы формально гидрид с формальной степенью окисления H(-1). Это не так просто, как может показаться. Хорошо бы использовать кислород, а ещё лучше воздух, но гидрид палладия слишком медленно окисляется кислородом - эта стадия стала бы бутылочным горлышком и очень узким, а гидрид палладия, если его оставить в растворе, довольно легко теряет протон, превращаясь в палладиевую чернь - на этом процесс окончательно бы остановился.
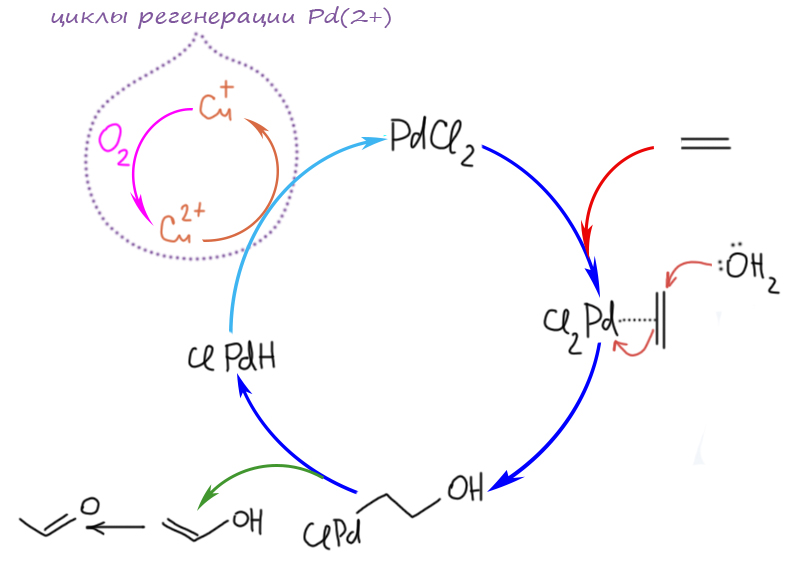
Но выход есть - для окисления гидрида можно использовать соль Cu(2+), тот же хлорид. И соль меди(1+) уже быстрее окисляется кислородом, поэтому и соль меди можно взять в каталитическом количестве. Цикл замкнут!
Некаталитический процесс стал каталитическим с помощью сопряжения с другим циклом - циклом регенерации Cu(2+). Чтобы всё это работало, разработчикам пришлось поломать голову, потому что всё это происходит с приемлемыми скоростями только в растворе довольно крепкой соляной кислоты - получилась настолько агрессивная среда, что в 1950-е не могли найти материала для реактора - ни одна специальная реакторная сталь не выдерживала. И тогда впервые решили попробовать титан, и этот металл выдержал. Кажется, это первый промышленный процесс, в котором нашёл применение этот потрясающий металл.
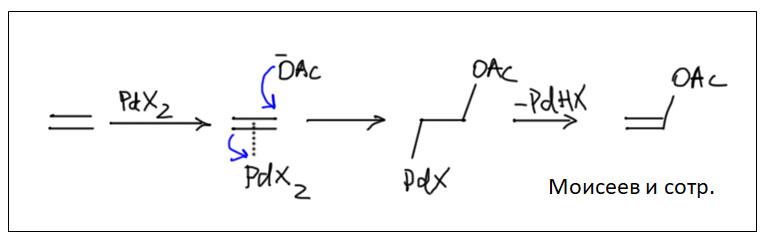
Кроме воды могут присоединяться и другие нуклеофилы. Такую реакцию открыл одновременно и независимо И.И.Моисеев с сотрудниками (Моисеев, И.И. и др. ДАН СССР, 1960,133, 377). Процесс Вакер-Хёхст был открыто описан и запатентован в 1959, и если вам кажется, что это раньше, попробуйте представить себе мир без интернета, в котором информация распространяется только на бумаге, и требуется несколько лет, чтобы она дошла до вас. Продукт этой реакции не менее, а даже намного более интересен, чем ацетальдегид - это винилацетат, важнейший мономер. Полимеры и сополимеры винилацетата - важнейшие компоненты современных красок, клеев, строительных компаундов. До изобретения этого метода винилацетат получали присоединением уксусной кислоты к ацетилену в присутствии кислот Льюиса, а следовательно имела те же проблемы с опасным и неудобным исходным, как и все другие ацетиленовые синтезы.

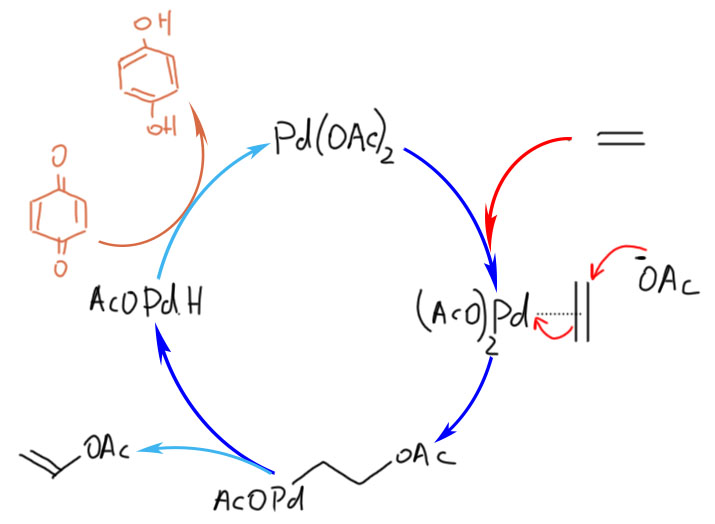
Реакция Моисеева каталитическая по палладию за счёт окисления гидрида палладия, но не медью(2+), а очень интересным органическим окислителем, бензохиноном. Бензохинон используется в стехиометрическом количестве. Открытие И.И.Моисеева было опубликовано в открытом научном журнале - в СССР просто не умели заниматься патентованием перспективных разработок, и не понимали, когда это нужно сделать. Поэтому его немедленно подхватили исследователи из той же компании Вакер, и довели до промышленного процесса, естественно заменив окислитель, потому что в промышленных условиях такой окислитель как бензохинон не имеет перспектив для крупнотоннажного процесса. В результате получился намного более удобный и экономичный газофазный процесс, в котором винилацетат образуется из этилена, уксусной кислоты на гетерогенном палладиевом катализаторе с использованием кислорода в качестве стехиометрического окислителя.
Как мы видим, превращение ацетилена в ацетальдегид (реакция Кучерова) - каталитическая, а превращение этилена в ацетальдегид (Вакер-процесс) - некаталитическая в отсутствие сопряжённого процесса регенерации активной формы палладия. Критерий различия очень прост - это стехиометрическое уравнение реакции. Если реакцию можно записать со стехиометрическими коэффициентами, то есть и шанс сделать её каталитической, подобрав катализатор, используемый в каталитических количествах.
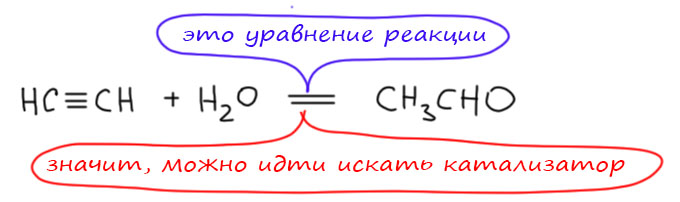
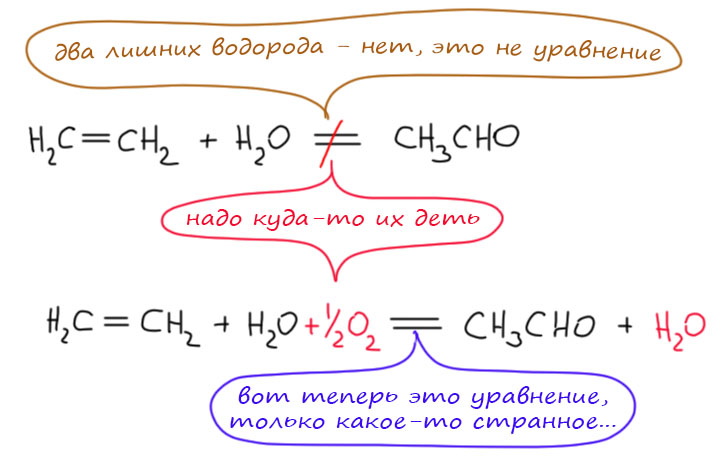
А вот превращение этилена и воды в ацетальдегид - это не реакция, она не уравнивается, в ней два лишних водорода
Поэтому ее нельзя просто сделать каталитической. Сначала мы должны добиться соблюдения требований Ломоносова и Лавуазье. Чтобы убрать водороды, нужен окислитель, например, кислород. Если добавим кислород, всё тут же уравняется. Это уже можно сделать каталитическим?
Не совсем и не сразу, ведь теперь в одном формально верном уравнении зашифровано два реальных - реакции этилена с водой с образованием какого-то интермедиата, и окисление этого интермедиата. Если мы можем соединить их в последовательность стадий, то получим каталитический цикл. Собственно, именно это и сделано в Вакер-процессе и реакции Моисеева.
Вакер-процесс в лаборатории использовать никто не будет, это чисто промышленная химия. А можно ли эту реакцию использовать в лаборатории не с этиленом, а с другими олефинами? Это был бы очень привлекательный метод синтеза кетонов из олефинов. Но прямо использовать условия Вакер-процесса не получится - они очень жёсткие, в них выживает только этилен, а все остальные олефины будут быстро или присоединять HCl или полимеризоваться. Решение нашёл в 1976 очень упорный и изобретательный японский химик Дзиро Цудзи, внёсший огромный вклад в палладиевый катализ в начале его развития. Мы с ним скоро ещё раз встретимся.
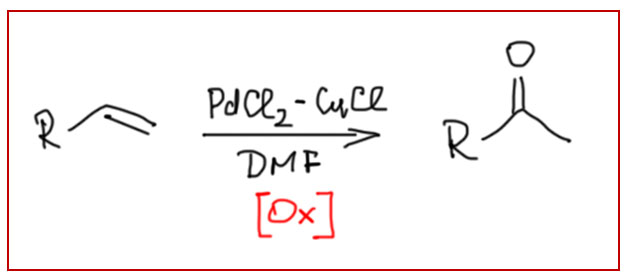
Применение Вакер-реакции для лабораторного синтеза неудобно, так как оригинальная тройная каталитическая система палладий-медь-кислород работает только в сильнокислой среде, неприемлемой для большинства синтезов сложных соединений. Дзиро Цудзи предложил очень привлекательный метод превращения терминальных олефинов в метилкетоны в более удобных условиях, в обычном органическом растворителе (ДМФА) и с использованием органического реокислителя гидрида палладия, в качестве которого стали использовать п-бензохинон. Метод получил название реакции Вакер-Цудзи, и получил дальнейшее и очень интересное развитие.
Донорный и акцепторный компоненты связи металл-двойная или тройная связь гораздо чаще не преобладают друг над другом, как в двух уже рассмотренных типах процессов, а находятся в тонком балансе, и в этом случае невозможно точно сказать, нуклеофильной или электрофильной является активация двойной или тройной связи, вошедшей в координационную сферу металла.
В этих случаях основной реакцией координированного олефина или ацетилена становится миграционное внедрение, происходящее по согласованному механизму в координационной сфере металла. Реакций, включающих стадии этого типа, огромное количество. Это и многочисленные реакции присоединения к кратным связям, в которых после миграционного внедрения металл замещается на какой-то другой фрагмент. Это и так называемые каскадные процессы, в которых стадия миграционного внедрения несколько раз повторяется, создавая сразу очень сложную структуру из достаточно простых исходных.
В близком родстве с миграционным внедрением находится реакция β-элиминирования. Мы очень хорошо знаем эту реакцию из обычной органической химии, где с ее помощью создают кратные связи. В химии переходных металлов все, на первый взгляд, очень похоже, но роль уходящей группы играет переходный металл, и именно он, а не внешнее основание эту реакцию инициирует. Стоит заметить, что реакция β-элиминирования не всегда играет положительную роль, и очень часто является причиной весьма неприятной побочной реакции, ведущей или к преждевременному выходу лигандов из координационной сферы, или к изомеризации олефиновых фрагментов и потере селективности.
Это наиболее распространенный тип превращения координированного олефина или ацетилена.
X-лиганд из координационной сферы металла мигрирует на один из атомов углерода, а η2-связанный алкен или алкин превращается в σ-связанный лиганд, фактически встраиваясь (внедряясь) по связи металл-мигрировавший X-лиганд. С точки зрения механизма это согласованный процесс, в котором разрыв и образование связей происходят одновременно, хотя и не обязательно синхронно. По этой причине становится неважно, каким типом реакционной способности - электрофильной или нуклеофильной - обладает "на свободе" мигрирующая группа X. Все делает металл, как такой манипулятор, берущий группу X одной рукой, то есть орбиталью, и пристраивающий ее на другой свой лиганд, который он держит другой рукой, то есть орбиталью, и возможно, не одной.
Стереохимический результат реакции - всегда син (цис), что отличает этот процесс от внешне очень похожего нуклеометаллирования, в котором нуклеофил приходит извне.
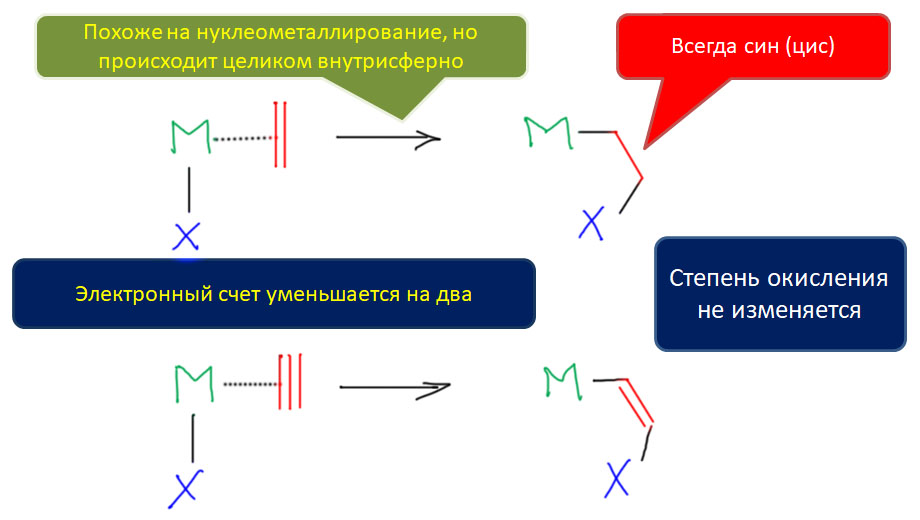
Реакции миграционного внедрения чрезвычайно распространены, и не ограничиваются соединениями с кратными связями. На них способны практически все переходные металлы - и ранние, и поздние.
Если миграционное внедрение - самый распространённый путь превращения олефина или ацетилена во внутренней сфере переходного металла, реакция β-элиминирования очень часто выводит продукт из координационной сферы. Эта реакция является обратной к миграционному внедрению, но гораздо более узкой. Эта реакция происходит только с достаточно электроположительными атомами и фрагментами. Самый распространенный вариант β-элиминирования - β-гидридное элиминирование, с которым мы уже несколько раз встречались. Для β-гидридного элиминирования необходимо син-расположение металла и атома водорода.
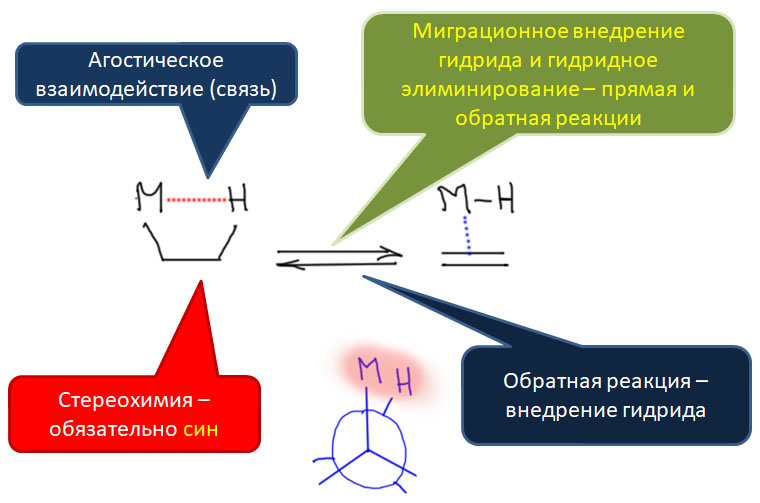
Процесс начинается с возникновения слабого взамодействия металл-водород (так называемой агостической связи) и развивается в четырехчленном переходном состоянии. После элиминирования алкен (алкин) остается в координационной сфере в виде η2-лиганда, но обычно бывает связан настолько слабо, что вытесняется из нее любым подходящим лигандом, например, фосфином или даже просто донорным растворителем. Поэтому β-гидридное элиминирование часто находится в конце каталитических циклов и отвечает за выход продукта из координационной сферы. В отличии от миграционного внедрения, гидридное элиминирование встречается далеко не у всех металлов, некоторые, как металлы 10 группы, его особенно любят, другие же предпочитают другие завершения своих манипуляций с лигандами. Тем не менее, это весьма общий путь, и найти его можно и у поздних, и у ранних переходных металлов.
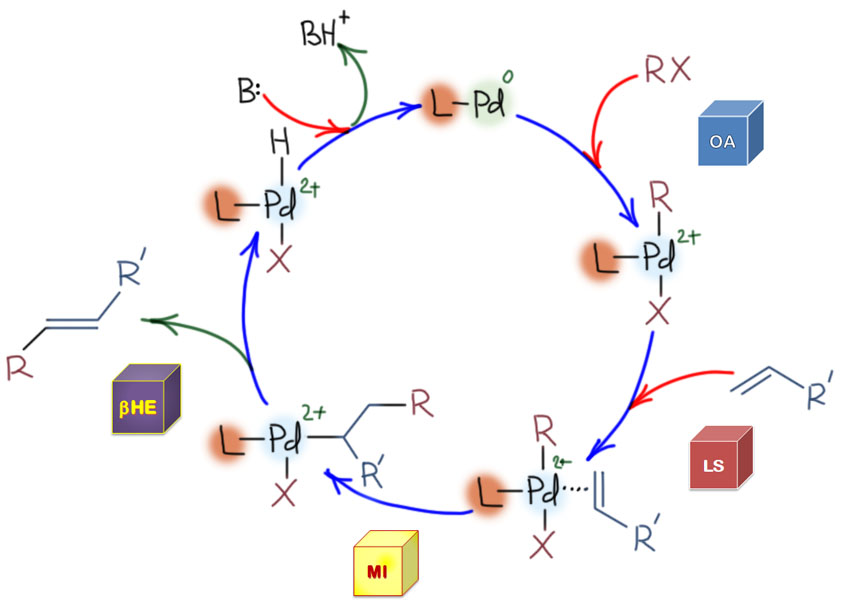
Последовательность окислительное присоединение – замещение лиганда – миграционное внедрение – восстановительное элиминирование встречается в десятках каталитических процессов. Типичным и одним из важнейших примеров таких процессов является гомогенное гидрирование. Сама по себе эта реакция очень важна для исследования каталитических процессов, но с точки зрения применения в синтезе не так интересна, потому что и так существует множество отличных катализаторов гетерогенного гидрирования, а при прочих равных условиях гетерогенный катализ удобнее. Но гомогенное гидрирование стало по настоящему важнейшей реакцией, когда был разработан его асимметрический вариант – получение оптически активных продуктов гидрирования из оптически неактивных олефинов с помощью хиральных катализаторов, в основном за счёт хиральных лигандов (возможна и хиральность на стереогенном атоме металла, и примеры таких комплексов и их применения есть, но это до сих пор довольно большая редкость).
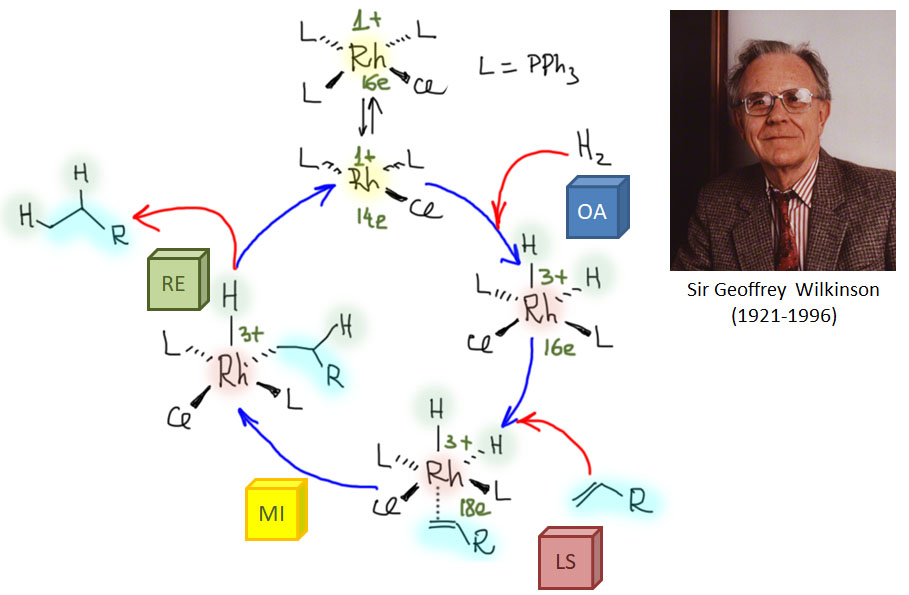
Первая эффективная реакция гомогенного каталитического гидрирования описана в работе (Wilkinson, J. Chem. Soc. (A), 1966, 1711) нобелевского лауреата Джоффри Уилкинсона и сотрудников (нобелевка не за это, а за ферроцен). Комплекс родия, получивший название катализатора Уилкинсона, способен в растворах быстро гидрировать многие олефины и ацетилены (и те, и те до алканов) с очень большими величинами TON/TOF. Процесс развивается в соответствии с типичным каталитическим циклов присоединения к кратным связям. В каталитический цикл входит координационно ненасыщенный комплекс родия (1+), который охотно окислительно присоединяет водород. Оба атома водорода последовательно переходят на атомы углерода, первый в результате миграционного внедрения, второй - за счет восстановительного элиминирования.
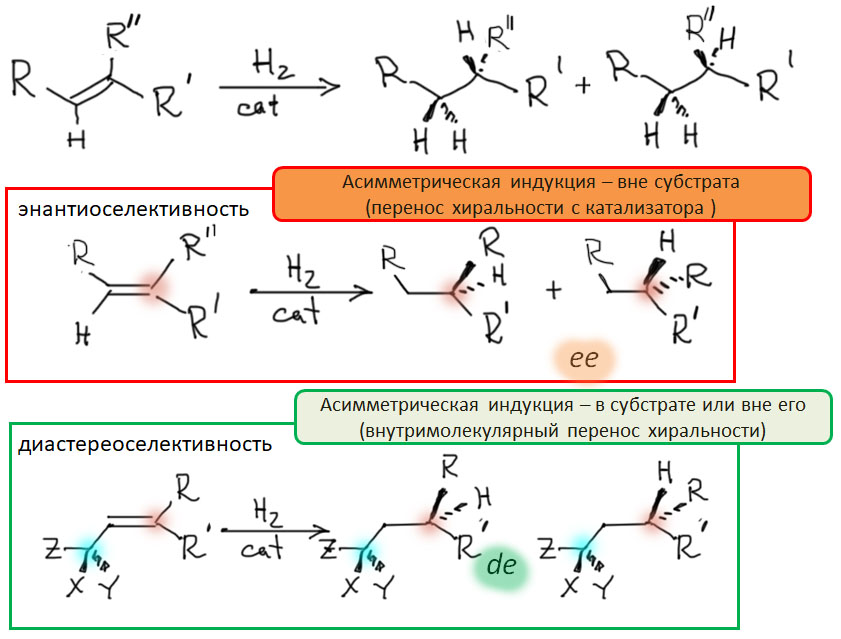
Характеристикой энантиоселективности является энантиомерный избыток (enatiomeric excess, ee) - это просто разность содержания в смеси энантиомеров того, которого больше, и того, которого меньше. Величина измеряется в % и изменяется от 0% для полной неселективности (в рацемате, 50%-50%=0%) до 100% при энантиоспецифической реакции (получился чистый энантиомер (100%-0%=100%).
Диастереоселективность характеризуется похожей величиной диастереомерного избытка (de)
Наиболее важным применением гомогенного гидрирования является стереоселективное гидрирование. Если при присоединении водорода один из атомов водорода присоединяется к атому углерода с двумя разными заместителями при двойной связи (про-хиральному атому углерода), то возможно, что продукт будет не рацемической смесью, а смесью энантиомеров с преобладанием одного. Если такой атом один, и в остальной части молекулы нет других стереогенных фрагментов, то реакция называется энантиоселективной, и источником преобладания одного из энантиомеров (асимметрической индукции) является катализатор гидрирования. Если в остальной части молекулы есть другие стереогенные центры или фрагменты, реакция называется диастереоселективной, и для такой реакции применение энантиоселективного катализатора не обязательно, хотя применение таких катализаторов и в этом случае может способствовать более высокой диастереоселективности.
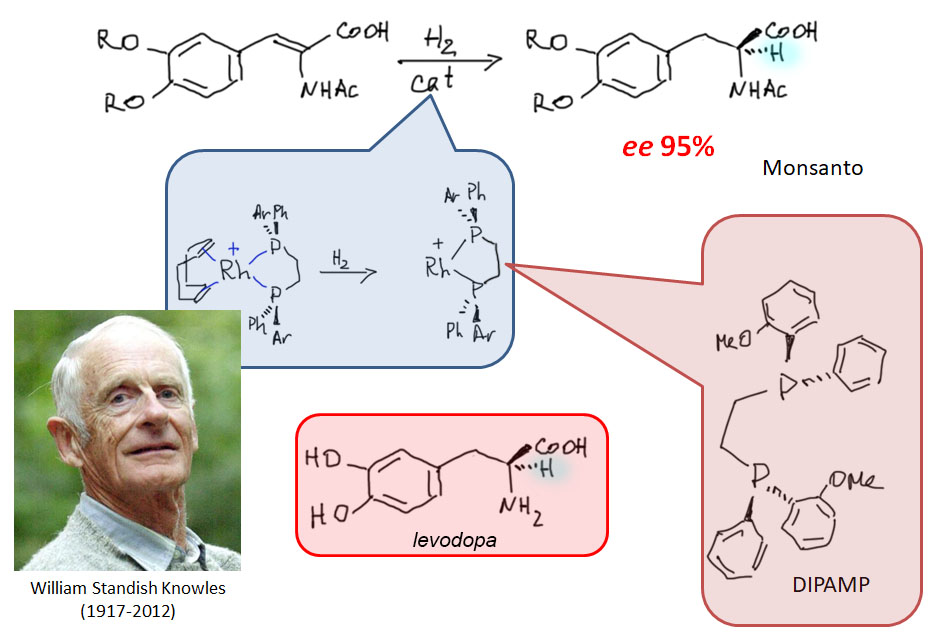
В 1970-х появился первый пример успешного применения энантиоселективного гидрирования. Уилльям Ноулз с сотрудниками разработали метод энантиоселективного гидрирования замещенной ацетиламинокоричной кислоты, с образованием непосредственного предшественника важного лекарственного препарата L-ДОФА (диоксифенилаланина) в энантиомерно чистой форме. Этот препарат успешно применялся и применяется до сих пор для облегчения симптомов паркинсонизма. Коммерческий успех метода подстегнул интерес к исследованиям энантиоселективного гидрирования, и с тех пор были разработаны методы синтеза не менее десятка важных лекарственных препаратов, включающих энантиоселективное гидрирование. Ноулз за эти исследования в 2001 получил Нобелевскую премию. В реакции используется катионный комплекс родия(1+) с бидентатным хиральным фосфиновым лигандом DIPAMP, оптическая активность которого обусловлена наличием двух хиральных атомов трёхвалентного фосфора в одинаковой конфигурации.
Причина высокой энантиоселективности гидрирования в синтезе леводофы - жесткое и надежное закрепления субстрата и хирального фосфина в координационной сфере родия. Исходный олефин для гидрирования связывается с металлом как через двойную связь, так и основным атомом азота в координационно-стабильный хелат, и именно это способствует стереохимически-точному образованию стереогенного центра в результате миграционного внедрения и восстановительного элиминирования.
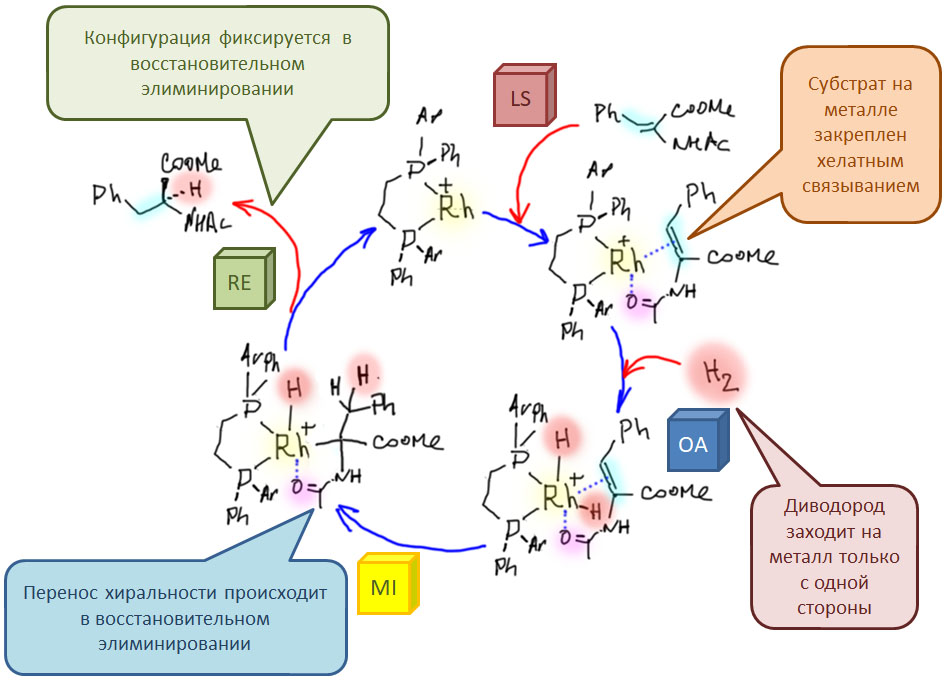
Проблема лигандов, хиральность которых обусловлена наличием стереогенных атомов (центров), типа того же DIPAMP состоит в их узкой специализации, - стоит взять другой субстрат, даже очень похожий, как результат становится непредсказуемым. Более универсальные лиганды, показывающие отличные результаты на очень широком круге реакций, получаются из более жестких хиральных молекул, имеющих более сильную асимметрию, а такие лиганды чаще всего делаются из молекул, имеющих единственный элемент симметрии - ось второго порядка. Самый знаменитый лиганд этого типа - BINAP - был предложен в группе Рёдзи Ноёри (именно так правильно, хотя чаще пишут Нойори). Рутениевые комплексы BINAP отлично работают в энантиоселективном гидрировании олефинов, но особенно популярны для энантиоселективного гидрирования карбонильной группы в оптически активные спирты.
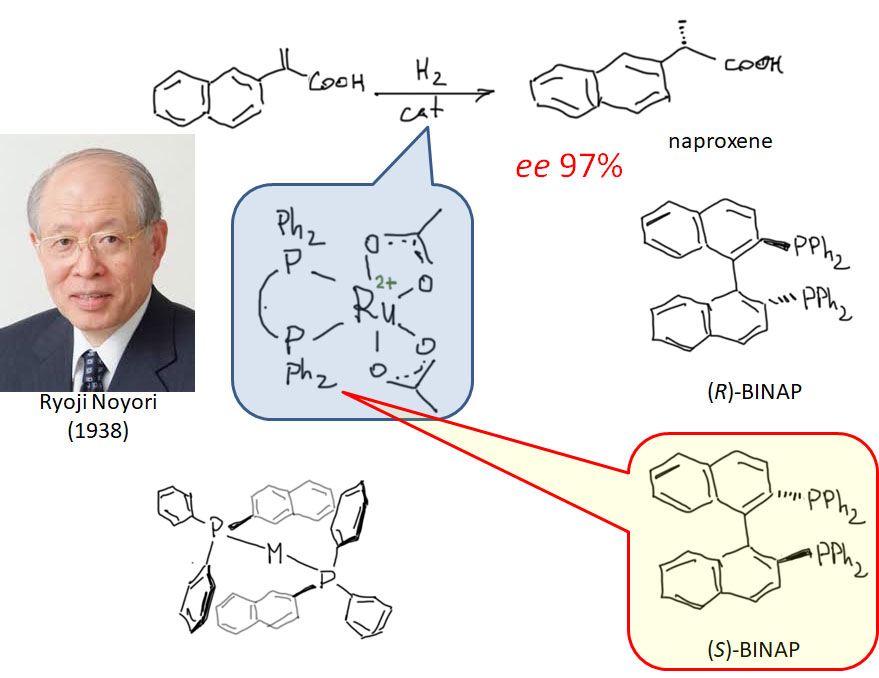
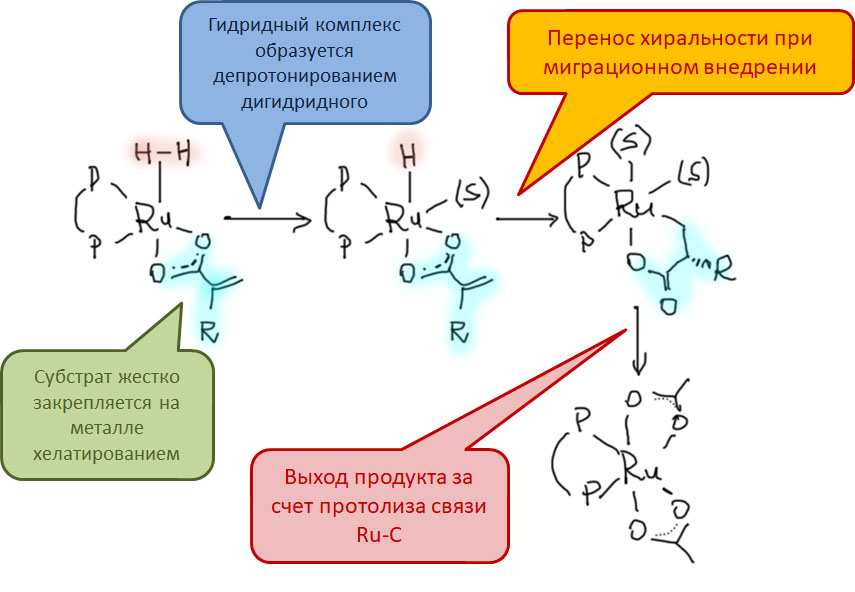
Гидрирование с рутениевыми комплексами BINAP протекает по совершенно другому механизму, который обычно называют моногидридным. Молекула водорода, связання в дигапто-комплекс, становится довольно сильной протонной кислотой и легко депротонируется слабым основанием с образованием гидридного комплекса. Тем не менее, основные черты каталитического цикла сохраняются: жесткое закрепление субстрата за счет хелатного связывания, перенос хиральности в миграционном внедрении.
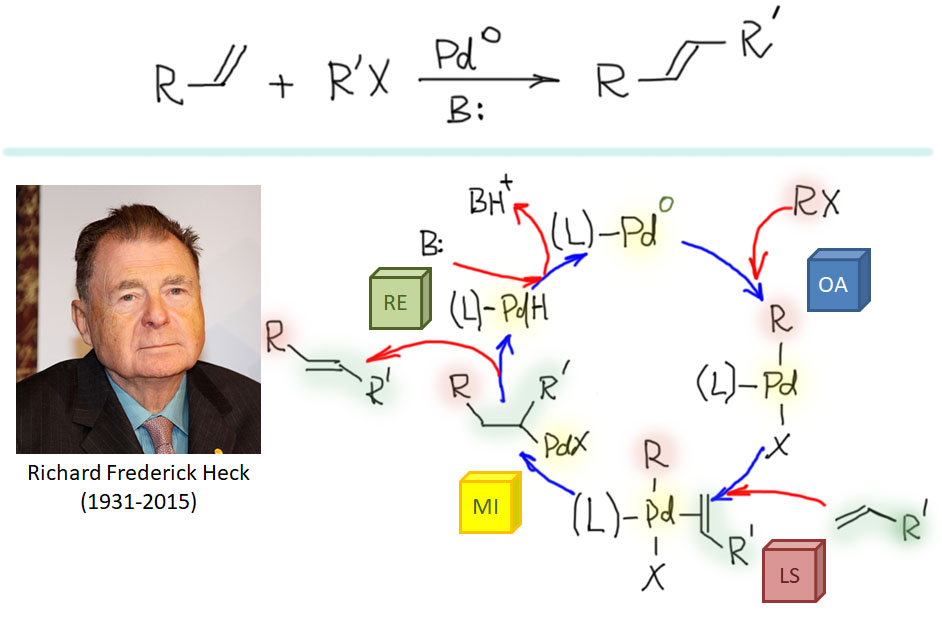
Но другой важнейший процесс описывается немного другим циклом, с другим завершением. Цикл окислительное присоединение – замещение лиганда – миграционное внедрение – β-гидридное элиминирование соответствует реакции Мидзороки-Хека. Но на этом история не кончается. В некоторых случаях реакция Мидзороки-Хека не может завершиться гидридным элиминированием по стереохимическим причинам, и тогда комплекс палладия с σ-органическим лигандом может принять участие в новых превращениях (еще одно карбапалладирование, всевозможные реакции кросс-сочетания, карбонилирование и т.п.), до тех пор пока каталитически активный комплекс палладия не будет регенерован для входа в новый цикл. Такие последовательности стадий происходят в рамках одного каталитического цикла и называются каскадами. Иногда еще используют термин domino reaction, имея в виду старинную забаву, когда костяшки домино ставят плашмя в длинный строй, и опрокидывая первую, вызывают лавинообразное заваливание всего построения. Когда костяшек много, это впечатляющее и незабываемое зрелище. Но я не люблю этот термин, считаю его избыточным (есть уже более точный и вполне определенный термин каскад), а что еще хуже, очень нечетким, потому что домино-реакциями иногда называют и тандемные процессы, а это уже терминологическая неразбериха и бардак.
Этот слайдер обновлён 17.04.2022. Новые или обновлённые слайды можно легко узнать по анимированным заголовкам и эффекту инверсии при наведении курсора на заголовок.
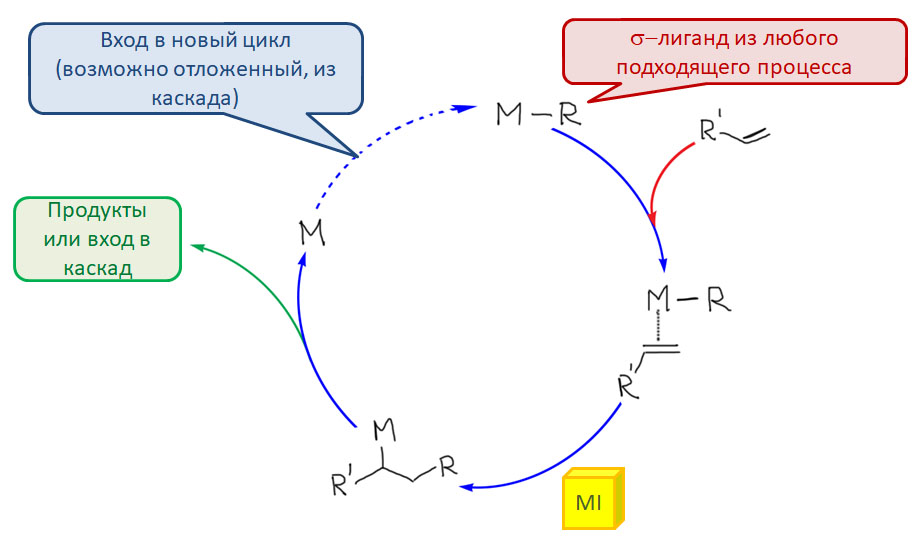
Миграционное внедрение сигма-связанного углеродного лиганда (алкила, арила, ацила и т.п.) с кратной углерод-углеродной связью, за которым следуют разнообразные превращения (восстановительное элиминирование, гидридное элиминирование, следующая стадия каскада) представляет собой исключительно мощный метод соединения углеродных фрагментов в новый скелет. Почти бесконечные возможности этой реакции определяют и разное поведение переходных металлов, и возможности управлять течением процесса за счет выбора лигандов, и потенциалом соединения этой реакции с предшествующими и последующими стадиями.
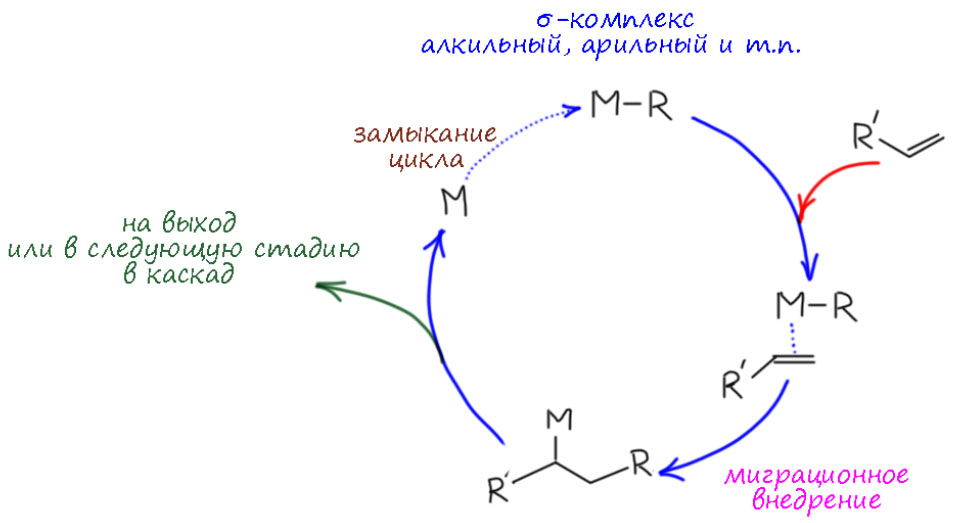
Карбометаллирование - одна из основных элементарных реакций, на которых строятся сложные каскады - большие каталитические циклы, включающие до десятка, а иногда и более, последовательных реакций.
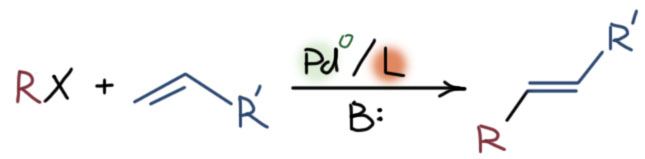

Ричард Хек
(1931-2015)
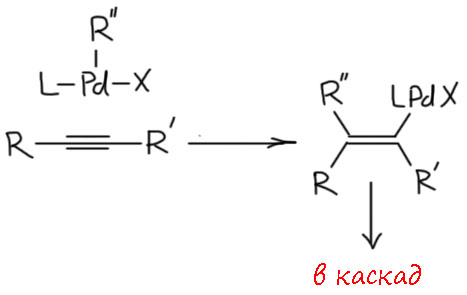
Важнейшая и самая популярная реакция, использующая карбопалладирование - реакция Мидзороки-Хека, открытая параллельно с классическими методами кросс-сочетания, и даже немного раньше. Ключами к открытию и кросс-сочетания, и этой реакции стали два достижения: окислительное присоединение галогенпроизводных к нульвалентным металлам 10-й группы, и разработка общих принципов каталитического цикла самим Хеком. В этом методе типичные органические электрофилы реагируют с олефинами с образованием новой C-C связи.
Реакция включает окислительное присоединение, вход олефина в координационную сферу, миграционное внедрение (карбопалладирование), и гидридное элиминирование. Регенерация нульвалентного палладия происходит за счет действия основания. В отсутствие основания реакция не является каталитической по палладию.
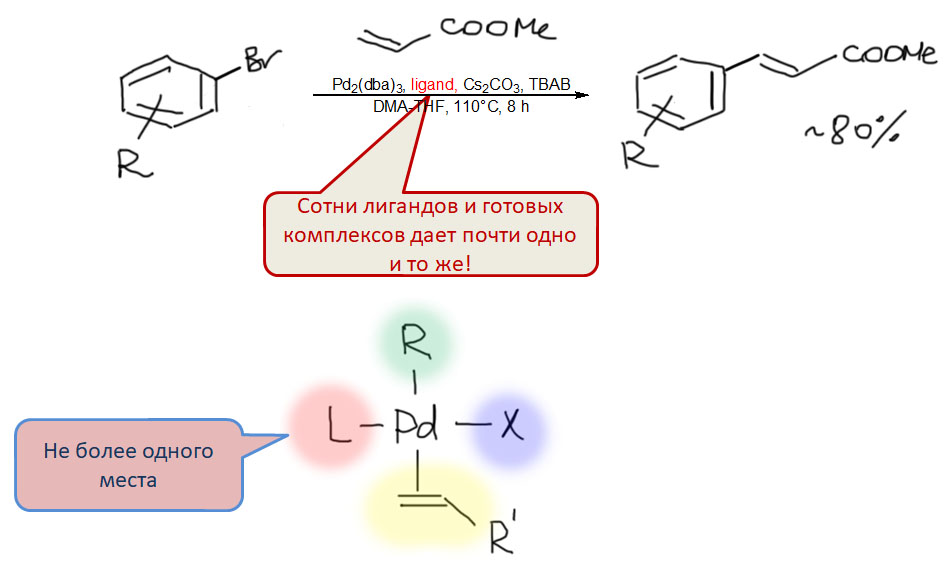
Реакция Мидзороки-Хека очень необычна тем, что на неё очень мало влияют лиганды и вообще структура пред-катализатора в том случае, если используются достаточно реакционноспособные бром и иодпроизводные.
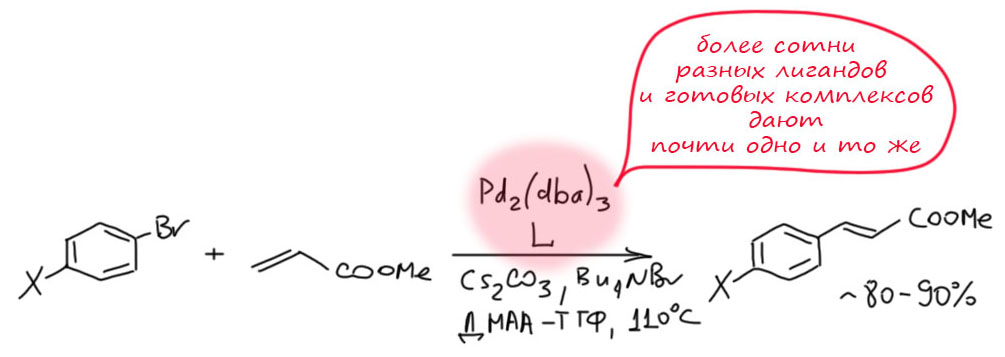
За почти четыре десятилетия очень интенсивных исследований накоплены огромные массивы информации по этой реакции, и эти горы данных в полном смысле этого слова родили мышь - вывод о том, что все эти исследования ничего не дали кроме этого парадоксального вывода.
Реакция Мидзороки-Хека формально очень похожа на кросс-сочетание, настолько, что ее часто причисляют к этому типу процессов. Но у реакции Мидзороки-Хека другой каталитический цикл (другой механизм), и другие требования к координационному окружению атома палладия. В реакции Мидзороки-Хека палладий должен располагать тремя доступными координационными местами у Pd(2+), которому свойственна плоскоквадратная конфигурация. По этой причине палладий не может иметь ни бидентатных лигандов, ни очень объемистых лигандов, а диапазон субстратов ограничн стандартными бромидами, иодидами, трифлатами.
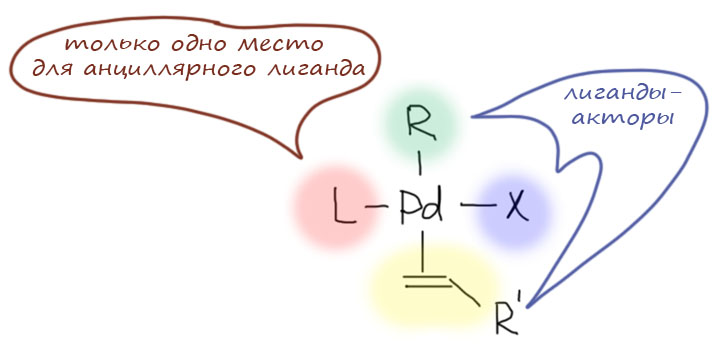
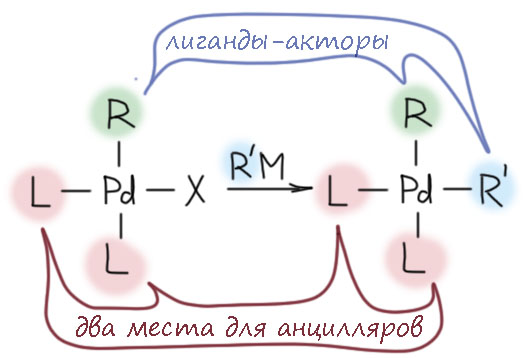
В кросс-сочетании на всех стадиях требуется не более двух мест для лигандов-акторов, потому что второй остаток входит через переметаллирование, использующее уже занятое место в координационной сфере. Поэтому в крос-сочетании активно используются бидентатные лиганды и специальные объёмистые и гемилабильные лиганды новых поколений. Это позволяет почти неограниченно расширять диапазон кросс-сочетания за счет сложных субстратов, плохих уходящих групп, необычных сред, и т.п.
Многие особенности реакции Мидзороки-Хека обусловлены очень строгой син-стереоспецифичностью двух ключевых реакций этого цикла - карбопалладированием и β-гидридным элиминированием
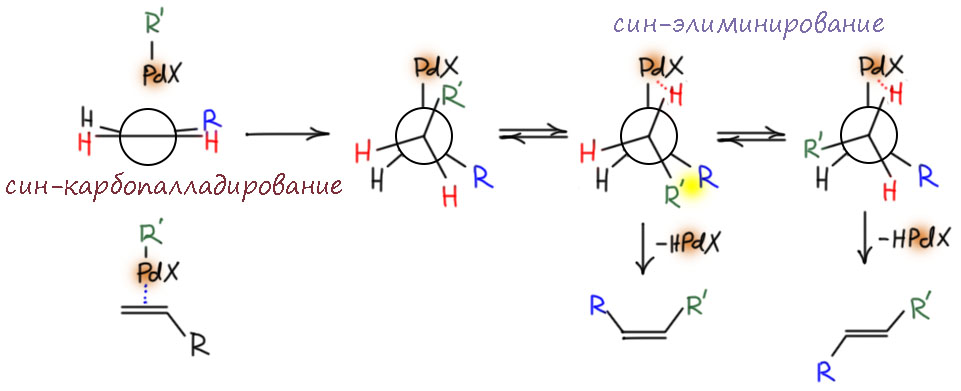
В реакции с терминальными олефинами обычно получаются транс-продукты (более 90% с небольшой примесью цис). Причина проста, и сразу видна из простого конформационного анализа. Так как и присоединение и элиминирование имеют син-стереохимию, из возможных конформеров более выгоден тот, который даёт транс из-за меньшего стерического отталкивания заместителей.
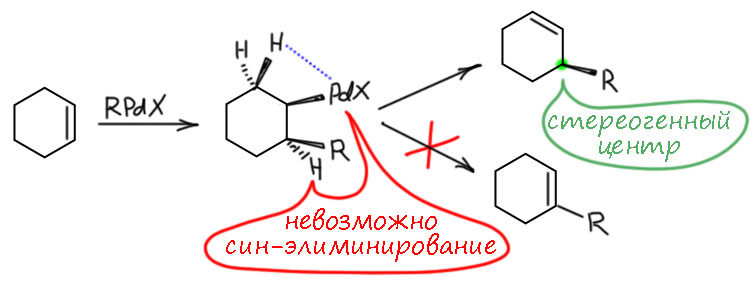
Син-син стереохимия объясняет и другие фокусы. Реакция с циклическими олефинами, например, не может дать обычный продукт реакции, так как невозможно син-элиминирование. Поэтому элиминирование происходит со следующего атома углерода в цикле, если такой есть, конечно, и если на нём есть подходящий водород. Это приводит к сдвигу двойной связи и заодно к образованию стереогенного центра, то есть создаёт возможность реализации энантиоселективной реакции Хека. Это, конечно, непросто, так как придется преодолеть то, с чем мы уже столкнулись - плохой контроль реакции со стороны анциллярных лигандов. Выход из этого есть, но об это в другой раз.
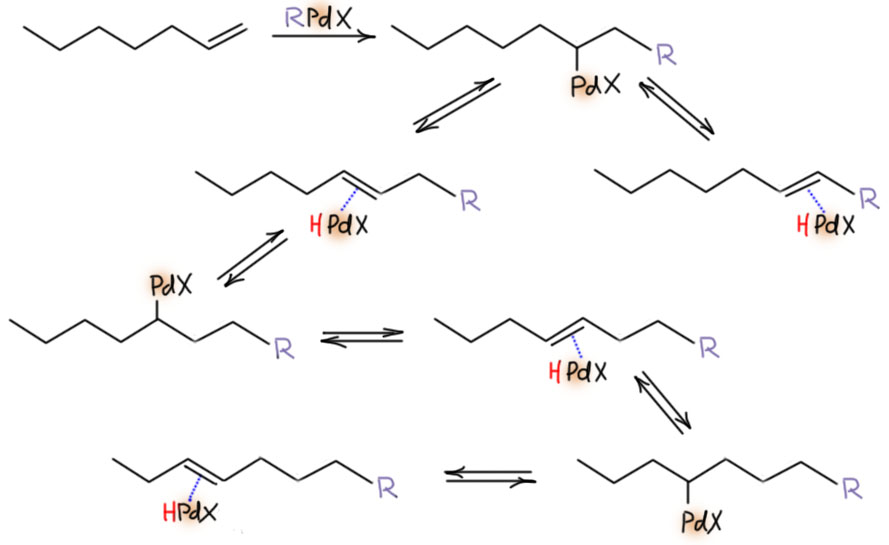
С нециклическими алкенами возникает другая проблема (или возможность). В этом случае гидрид для элиминирования есть и стереохимически доступен по обе стороны от металла, и элиминирование может происходить в обе стороны. Более того, элиминирование гидрида - обратимая реакция (обратная реакция - миграционное внедрение или гидропалладирование), и повторение этих стадий приводит к расползанию двойной связи по доступной части углеродной цепи. Как правило, это приводит к осложнениям, низкой селективности, и прочим плохим последствиям, но в некоторых случаях может быть использовано для интересных превращений.
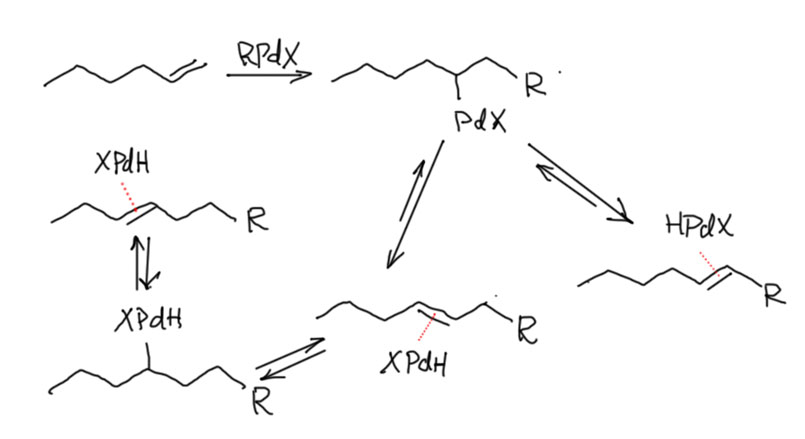
В этом процессе очень много зависит от конкретных условий, так как мы имеем дело с конкуренцией двух реакций - депротонирования гидрида палладия, и обратного присоединения (гидропалладирования) к двойным связям. Это всегда очень трудно предсказывать и еще труднее - пытаться управлять, поэтому мы в большинстве случаев имеем неустановившееся и очень подвижное равновесие с невоспроизводимыми результатами. Теоретически, это можно было бы использовать для смещения двойной связи, что-то типа ацетиленовой молнии (зиппер-реакции), но для этого нужно придумать, как создать один единственный продукт, выводящий систему из равновесия. Для палладиевой химии это сделать очень непросто, но аналогичные и вполне работающие системы есть в химии других переходных металлов.
Бидентатные лиганды, фосфины типа dppf или BINAP все-таки можно использовать в реакции Мидзороки-Хека, но только, если электрофилом является трифлат (есть и ещё несколько вариантов, например, соли диазония, но это экзотика). Теоретически можно представить три разных пути реакций с участием комплексов палладия с бидентатными лигандами: гемилабильный, неполярный и полярный. Несколько десятилетий поисков каталитических систем для реакций Хека выявили то, что только полярный путь является работающим, и именно таким способом были реализованы разные специальные разновидности реакции Мидзороки-Хека типа энантиоселективной.
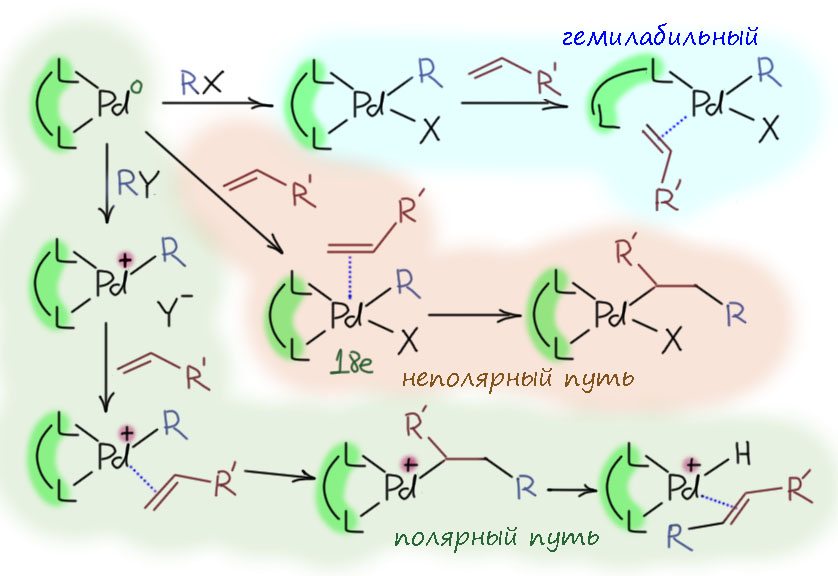
Гемилабильный хелатор мог бы уступать одно место для олефина, но в химии реакции Хека такие каталитические системы достоверно не известны
Олефин может связываться через 5-координированный 18-электронный интермедиат. Такой путь обычно называют неполярным, и он принципиально возможен, хотя дальше по циклу возникнет еще одна проблема с бета-гидридным элиминированием.
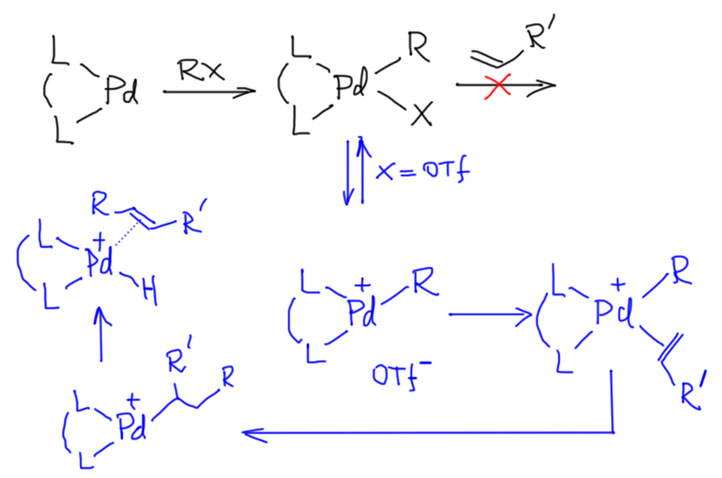
Наиболее реалистичный и, скорее всего, основной или даже единственный реальный путь связан с особым типом субстратов, содержащих высоконуклеофугные группы типа трифлата. Такой путь называют полярным.
Трифлат почти никогда не является лигандом в реакциях с участием поздних переходных металлов, и не занимает места в координационной сфере, освобождая его для реагирующих лигандов. Окислительное присоединение к трифлатам идёт как нуклеофильное замещение, так как трифлат ведёт себя, как и положено трифлату, как хорошая уходящая группа. Реакцию Мидзороки-Хека с трифлатами поэтому используют для специальных целей, например, для энантиоселективного арилирования циклических олефинов.
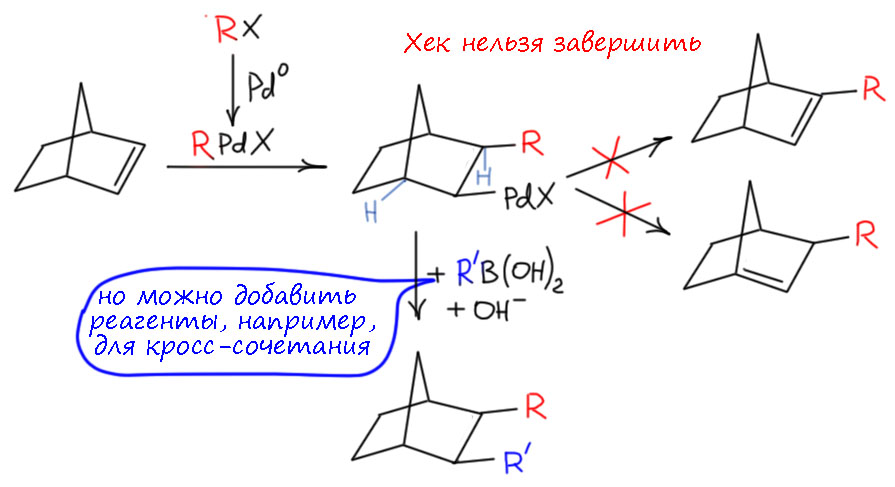
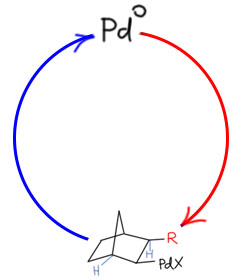
Бывает, что гидридное элиминирование совсем не может произойти, так как ни с одной стороны нет атома водорода. Самый знаменитый олефин такого типа - норборнен, подробно изученный в работах Марты Кателлани и сотрудников. После карбопалладирования реакция как будто зависает на палладий-органическом интемедиате. И можно дать ему возможность двигаться дальше, но не по пути Хека, а, например, по пути кросс-сочетания. Такую последовательность реакций, в которой интермедиат в каталитическом цикле перехватывается другой реакцией, называют каскадом.
Каскад - это единый каталитический цикл, а не последовательность из нескольких циклов или что-то ещё. Каскад обслуживается одним катализатором и имеет одну точку входа и одну точку выхода, в которой катализатор возвращается на новый виток. TON/TOF считаются по всему циклу, а не по каждой реакции каскада отдельно (тем более что реакции эти неразделимы). Каждая часть каскада в свою очередь может состоять из нескольких элементарных реакций, то есть представлять собой часть более простого каталитического цикла (реакции Хека, кросс-сочетания, гидрирования, и т.п.), по какой-то причине или незаконченного, или начинающегося не с обычного начала. Эти части склеиваются в большой цикл-каскад на общих интермедиатах, вот как здесь - интермедиат, образовавшийся в незаконченном Хеке, входит в кросс-сочетание на стадии переметаллирования.
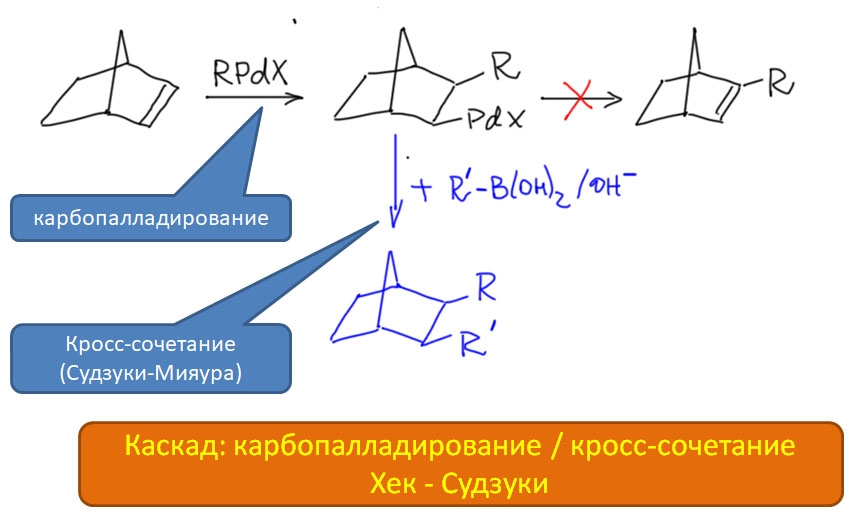
Еще более сложные каскады строятся на основе внутримолекулярных реакций. Готовится субстрат, содержащий набор реакционных центров, которые могут участвовать в последовательных реакциях карбопалладирования без возможности гидридного элиминирования, и таким образом, чтобы последовательно замыкались 5-ти или 6-тичленные циклы. В конце паллаиевый центр глушат какой-нибудь подходящей реакцией, замыкающей каскад.
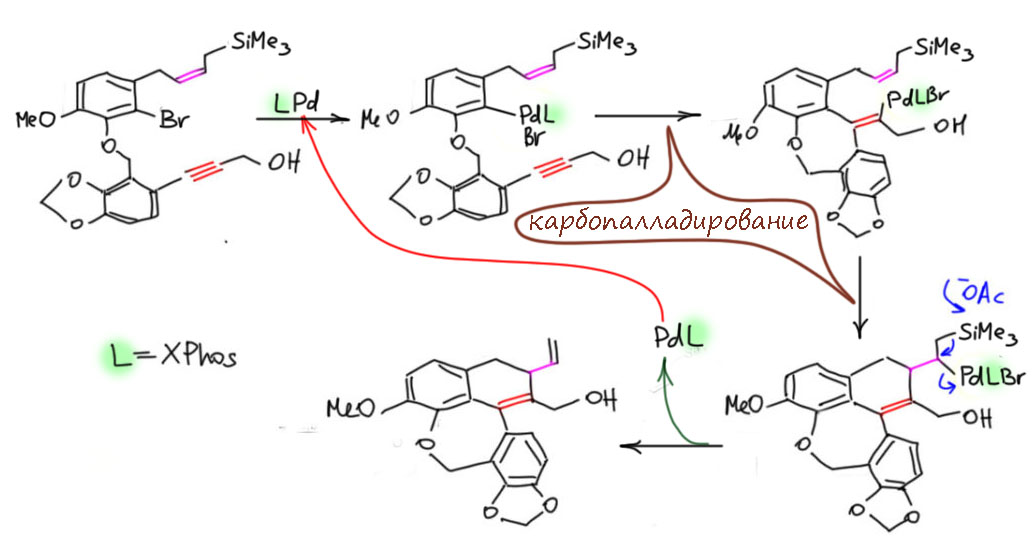
Очень удобны для этого не двойные, а тройные связи, при карбопалладировании которых принципиально не может получится структура, в которой возможно гидридное элиминирование. Поэтому в межмолеккулярных реакциях алкины используют редко, так как не всегда удается погасить палладиевый центр и начинается тупая олигомеризация. Но во внутримолекулярных реакциях использовать тройные связи - одно удовольствие, так как у них нет никаких неконтролируемых побочных.
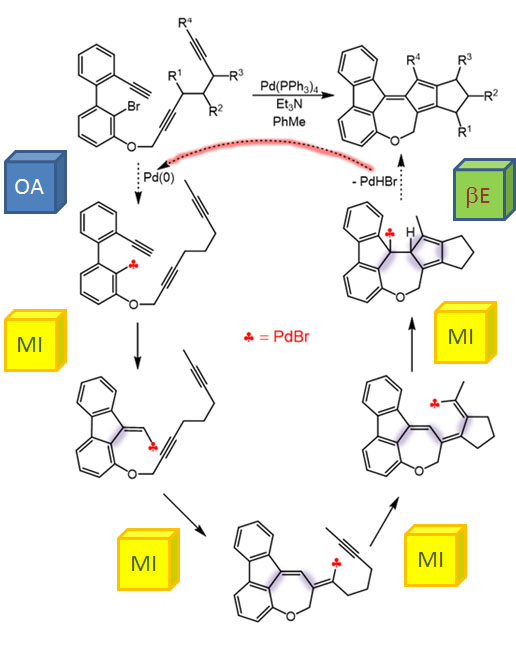
Вот один такой каскад из работ выдающегося каскадёра Луца Тице (L.Tietze et al. Angew. Chem. Int. Ed. 2013, 125, 3273) - два карбопалладирования с необычным завершением элиминированием силильной группы - в один присест, в одном каталитическом цикле создается сложная система циклов. Подобнее этот каскад разобран на страничке с Вопросами.
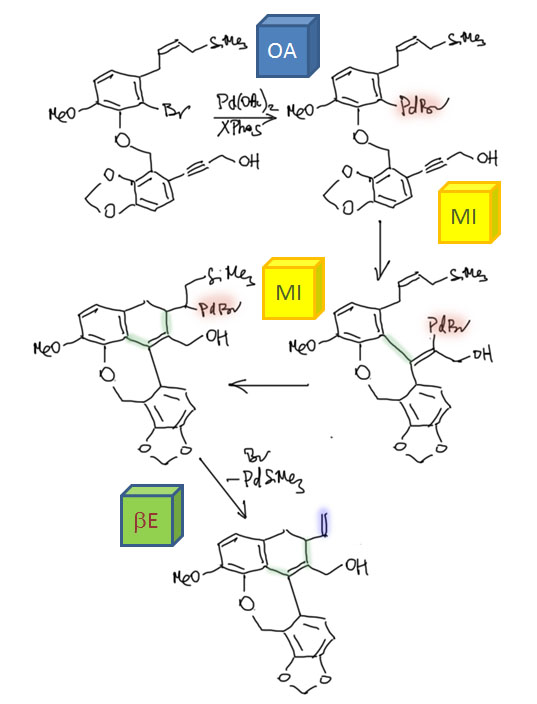
Еще более сложный каскад состоит из четырех последовательных карбопалладирований, причем последнее происходит на двойную связь, сформировавшуюся на первом карбопалладировании, которой не было в исходном, то есть надо было просчитать все ходы до последнего. В последнем карбопалладировании возникает неподходящий по конфигурации атом водорода для син-гидридного элиминирования, цикл всё равно завершается. Как это может произойти разобрано на отдельной страничке по кнопке Подробнее. Поэтому этот замечательный каскад можно считать таким четверным Хеком, поиском подходящего атома водорода для элиминирования, для чего пришлось поназамыкать кучу циклов в последовательности ходов, напоминающем изощрённую шахматную комбинацию. В каскаде из нециклической структуры (бензольные кольца каскадом не затрагиваются) образуется четыре новых кольца, три пятичленных и одно семичленное, причем одно из новых колец имеет фульвеновую структуру, а вся система является пентафульваленом.
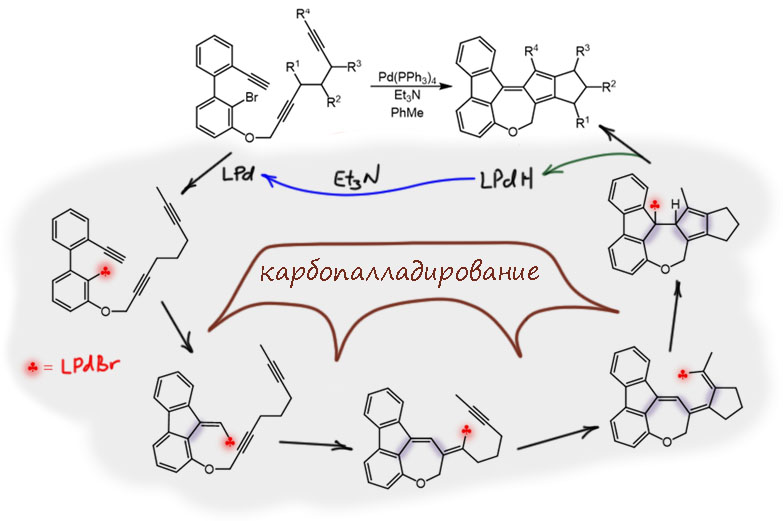
И для такой потрясающей каскадины не понадобилось ничего особенного - только самая банальная каталитическая система для простейшего Хека с самым тривиальным лигандом, да это и понятно, более крупный современный линганд, скорее всего, застрял бы на одной из первых стадий. Мораль - не выбрасывайте баночку с трифенилфосфином даже если накупили всех наисовременнейших лигандов и поддались на рекламу их чудодейственных качеств.