
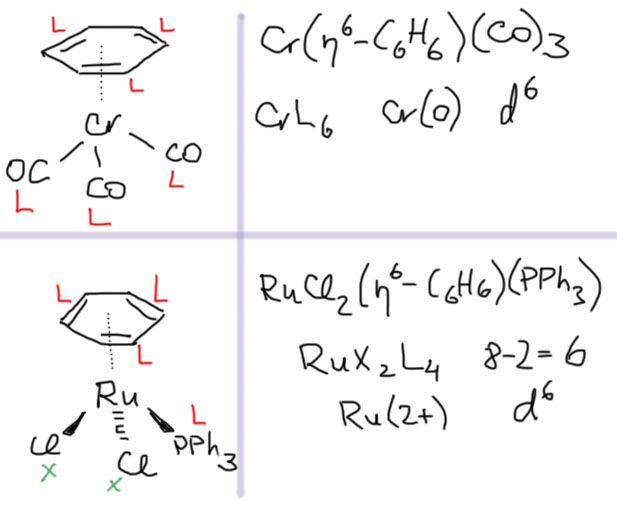
Связь как лиганд
Теперь можно сделать еще один шаг. Как мы видели, связь металла с лигандом обеспечивается парой электронов и в лигандах L-типа и в лигандах X-типа, при этом предполагается, что металл связан с монодентатным лигандом через один донорный атом, а с полидентатным лигандом – через n атомов, соответственно его реальной дентатности.
Но у d-элементов есть и более интересная возможность: они могут зацепить молекулу не через атом, а через связь, и ничего странного здесь нет. Сразу скажем, что заголовок этой главы, конечно, немного некорректен, потому что связь не является лигандом – лигандом является вся молекула, в которой находится эта связь. Связь – это координационный центр, в том же точно смысле, в котором таким центром мы считаем атом с неподеленной парой. Металл ищет у потенциального лиганда пару доступных электронов. Любая связь обслуживается как раз парой и почему бы ее и не подцепить. Безусловно, это не так просто как атом с нормальной несвязевой парой, так как связевые пары они на то и связевые, что имеют обычно более низкую энергию, а следовательно и менее доступны. Поэтому собственно мы раньше и не видели ничего подобного в химии непереходных элементов (а точнее, все-таки видели, но не обращали особого внимания: намекну, что что-то подобное можно увидеть, во-первых, в химии метониевого иона, и во-вторых, в так называемых мостиковых ионах в электрофильном присоединении к двойной связи – мы еще вернемся к этой далекой аналогии позже).
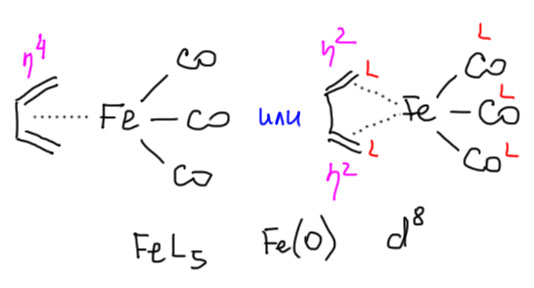
Здесь придется немного порисовать орбитали, в самом простом виде, без подробностей. Сначала попробуем подцепить простую одинарную связь: возьмем две молекулы, водород и бромистый метил. И там, и там простая σ-связь обслуживается связывающей орбиталью, в молекуле водорода она образована двумя s-орбиталями с одним знаком, в связи C-Br – двумя гибридными (со стороны брома это просто p-орбиталь) с одинаковым знаком перекрывающихся долей.
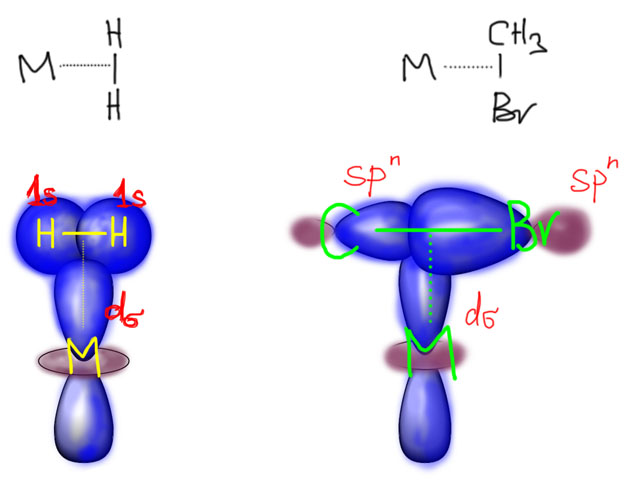
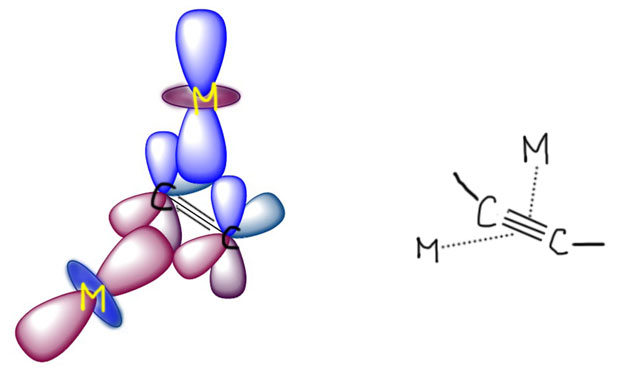
И что получилось? Ответ прост, типичная трехцентровая связь, пара электронов от σ-связи лиганда, пустая d-орбиталь от металла. И, обратите внимание, новая связь в комплексе – это тоже именно σ-связь. А почему на картинке написано d-сигма? Остановимся ненадолго на этой важной характеристике орбиталей и связей – сигма или пи?
Координационные σ- и π-связи
Понятие “σ- и π-связь” совершенно одинаково что в органической химии, что в координационной. Различить их очень просто, если мы имеем представление об орбиталях, которые обслуживают связь под вопросом. Делаем так:
- Проводим линию связи. Если связаны два атома, то это совсем просто – соединяем их (центры). Если атом связан с другой связью, то проводим через атом и середину этой связи. В более сложных случаях, а такие будут, простые советы не работают, но мы и не будем в таких случаях волноваться по поводу типа связи.
- Изображаем орбитали, обслуживающие связь.
- Если линия связи бежит по одной фазе (одному знаку, одному цвету) – то это сигма-связь.
- Если линия связи бежит по узловой плоскости (промежутку между долями орбиталей, между цветами) – то это пи-связь.
- Третьего не дано.
Теперь смотрим на картинки в предыдущем слайде. Видим, что в обеих случаях линия связи бежит “по синему” – по одному знаку или фазе орбиталей, образующих координационную связь. Решено – это именно сигма-связь и в первом и во втором случае.
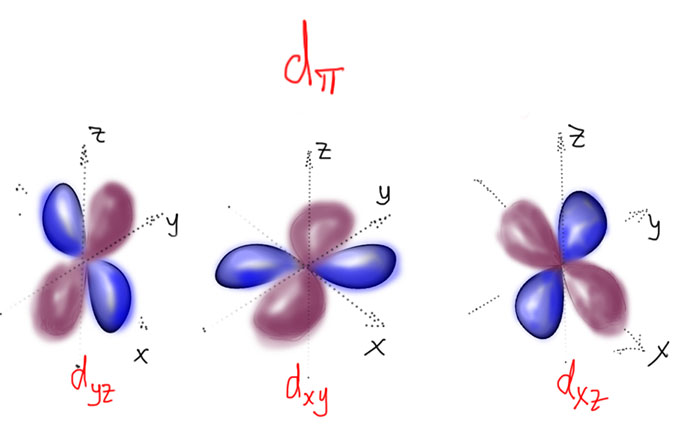
Вроде бы все просто, но остается вопрос, а как выбирать правильные d-орбитали – они ведь, в отличие от s- и p- разные. Здесь все довольно просто. D-орбитали действительно разные, но образуют они две группы: первые годятся для образования сигма-связей и называются dσ, вторые – для пи связей и называются dπ. D-сигма орбитали направлены вдоль осей координат. Каких таких осей координат, и каким образом они знают, где находятся лиганды?! Вот лиганды и определяют направление осей координат – мы их так и будем проводить, чтобы они проткнули максимальное количество лигандов. Особенно хорошо это получается в октаэдрических комплексах, и любых других, полученных из октаэдра обрезанием вершин (пирамида, квадрат, Т-треугольник, линия). Но и в других коодинационных геометриях проблем не будет. И не забывайте, что выбор орбиталей вполне произволен, так что всегда можно под интересующий нас лиганд “подогнать” самую удобную “орбиталь-с-юбочкой” (на картинках с предыдущего слайда мы так и сделали), но как только мы зафиксировали одну орбиталь, все остальные автоматически встанут на свои места (точнее, следующая орбиталь сохранит свободу в плоскости xy, но после нее все уже совсем однозначно).
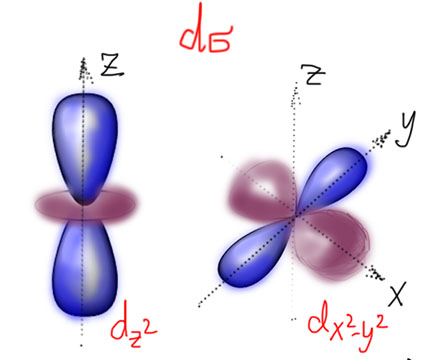
Три оставшиеся орбитали сидят между осей, каждая в своей плоскости, которая и определяет обозначения.
Комплексы олефинов и ацетиленов: σ-связь с π-связью!
 Возвращаемся к комплексам металлов, в которых донором пары является не атом, а связь. Очень легко образуют такие комплексы олефины. Настолько легко, что соль Цайзе (Zeise), K[PtCl3(C2H4)]·H2O – вообще считается едва ли не первым металлоорганическим соединением. Это соединение было случайно получено в 1830 году датским химиком Вильямом Цайзе, когда и химии-то еще никакой практически не было, при живом Пушкине, когда этот исключительно внимательный естествоиспытатель, впечатлённый реакцией спирта с хлором, решил вместо хлора взять хлористую платину. Потребовалось больше ста лет, чтобы разобраться, как устроена соль Цайзе. Вот она тут слева крутится – обычный плоскоквадратный комплекс платины(2+) (структура: Бокий Г.Б., Кукина Г.А. Ж.Структ.Хим. 1965,6, 706).
Возвращаемся к комплексам металлов, в которых донором пары является не атом, а связь. Очень легко образуют такие комплексы олефины. Настолько легко, что соль Цайзе (Zeise), K[PtCl3(C2H4)]·H2O – вообще считается едва ли не первым металлоорганическим соединением. Это соединение было случайно получено в 1830 году датским химиком Вильямом Цайзе, когда и химии-то еще никакой практически не было, при живом Пушкине, когда этот исключительно внимательный естествоиспытатель, впечатлённый реакцией спирта с хлором, решил вместо хлора взять хлористую платину. Потребовалось больше ста лет, чтобы разобраться, как устроена соль Цайзе. Вот она тут слева крутится – обычный плоскоквадратный комплекс платины(2+) (структура: Бокий Г.Б., Кукина Г.А. Ж.Структ.Хим. 1965,6, 706).
В комплексах двойной связи с переходными металлами донором пары является связывающая орбиталь двойной связи. С алкинами то же самое, только там π-связей две, и возникают дополнительные возможности для связывания. Это, как мы отлично знаем, настоящая π-связь. Легко в этом убедиться еще раз, если провести линию между атомами углерода, она побежит как раз в узловой плоскости (между синеньким и красненьким, если обозначать знак или фазу орбитали цветами). Но поскольку у такой орбитали при подходе к ней сбоку, к центру между атомами углерода, фаза (знак, цвет) с одной стороны одинаковые, то перекрывание опять обеспечивается одной долей dσ-орбитали, и связь с металлом оказывается поэтому настоящей σ-связью (проведем линию через атом металла и середину двойной связи – она побежит по одному цвету, “по-синенькому”. Вот она, первая гримаса координационной химии! Комплекс π-связи с металлом образуется σ-связью.
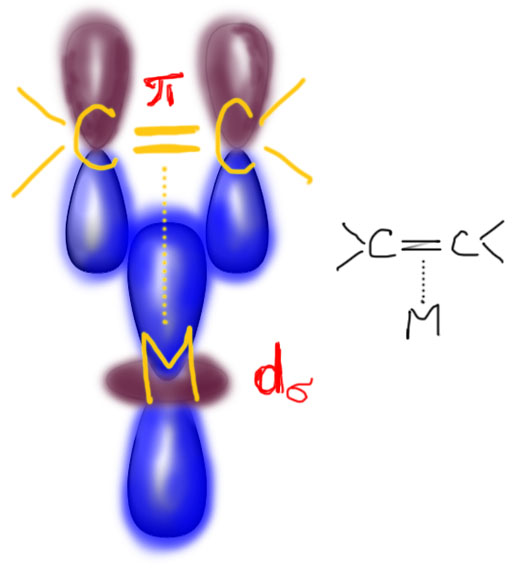
Неприятности на этом не кончаются. Не у нас, а у координационной химии. Дело в том, что комплексы металл-связь в химии испокон веков принято называть π-комплексами, и мы даже употребляли этот термин в курсе органической химии, когда обсуждали механизм электрофильного присоединения к олефинам, но особенно не вдаваясь в подробности. И в координационной химии такие комплексы называют π-комплексами, чтобы отличать их от комплексов со связью металл-атом, называемых σ-комплексами. Получается, что π-комплексы, по крайней мере в случае, когда лиганд связан с металлом не через атом, а через связь, образованы σ-связью, ничем принципиально не отличающейся от σ-связи в комплексах с простыми лигандами. Координационная химия, увы, не может похвастаться безупречной терминологией, и путаницы это вызывает немало. Будем бдительны!
Как учитывать лиганды, связанные с металлом через связь и как их покороче называть
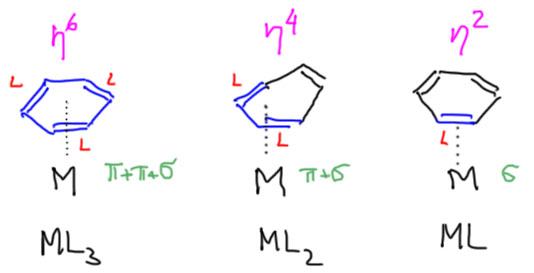
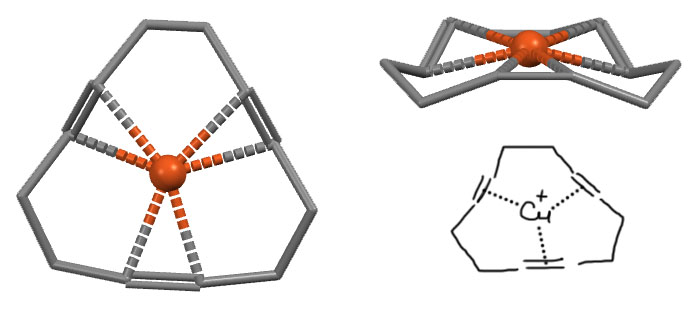
Кажется, что лиганды этого типа связаны с металлом не одним, а сразу двумя атомами, так как расстояние от атома металла до обоих обычно достаточно мало – получается такой треугольник (кстати, не обязательно равнобедренный – несимметричная связь между сильно разными атомами может немного скособочиться). Формально, такие лиганды так и рассматриваются – металл считается связанным с обоими атомами координированной связи. Для обозначения такой ситуации используется довольно корявое слово – гаптность (hapticity), а лиганды называются гапто-лигандами, от греческого слова, означающего “касаться, трогать, быть связанным”. Если металл кажется связанным с двумя атомами лиганда, такой лиганд называют дигапто-лиганд. Пока мы других и не знаем. Обозначается это греческой буквой эта, η с числом в надстрочнике – η2. Например, η2-диводород, η2-этен, η2-этин.
Каждый такой лиганд считается лигандом L-типа. Одним лигандом! Это хорошо видно по тому, что комплексы с гапто-лигандами имеют те же самые структурные типы, что и комплексы с простыми лигандами. Например, комплекс серебра с η2-ацетиленами (A.Reisinger, N.Trapp, I.Krossing, S.Altmannshofer, V.Herz, M.Presnitz, W.Scherer, Angew.Chem.,Int.Ed. 2007, 46, 8295) изоэлектронен и изоструктурен с часто встречающимися в химии серебра и других металлов 11 группы тетраэдрических комплексов с донорными L-лигандами с конфигурацией d10, только у ацетиленового комплекса в вершинах тетраэдра не атомы, а геометрические центры тройных связей, из которых формально растут формальные линии координационных связей.

Оговорюсь, что из этого правила есть исключения, и мы их когда-нибудь обсудим. Пока – без исключений. Посмотрим на примеры. Обратите еще внимание, что в одном комплексе водородные лиганды могут быть разными.
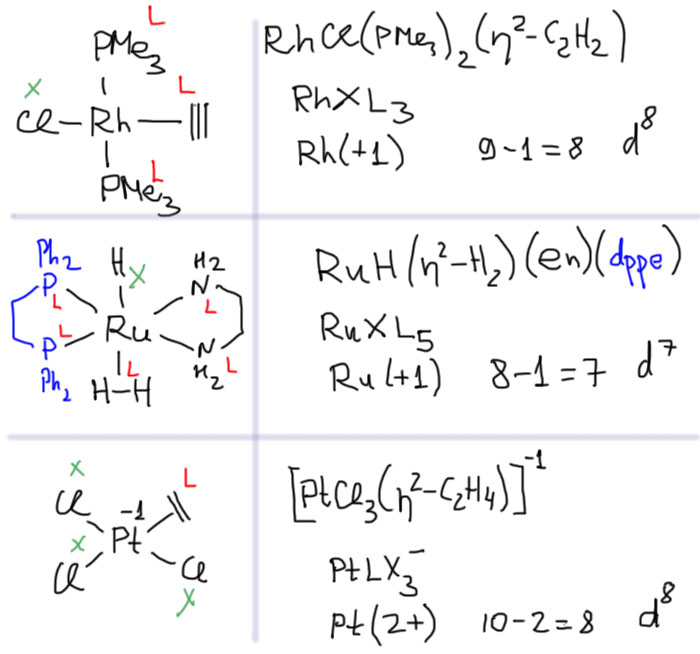
В случаях, когда в одном комплексе есть лиганды разных типов, иногда непросто понять, как это устроено. Особенно непросто это бывает, когда лиганды – это водород, который почти невозможно однозначно добыть из рентгеноструктурных данных. Тогда на помощь приходят общие соображения и сведения по реакциям таких спорных комплексов. С реакциями разберёмся позже. Посмотрим на общие соображения. Вот есть комплекс, по составу гексагидрид рутения с двумя фосфиновыми лигандами RuH6(PR3)2. Что это, действительно комплекс Ru(6+)? Ничего невозможного в этом нет, у рутения есть такая степень окисления, не сказать, что очень популярная, но есть. Но это будет комплекс с координационным числом 8. Вот это уже сомнительно, тем более что два лиганда довольно объёмистые. Аналогов не видно. Вывод – не невозможно, но очень маловероятно. Другая гипотеза – выводим часть водородов в молекулы водорода. Сколько? Два атома в одну молекулу – будет комплекс Ru(4+) с к.ч. 7 – очень сомнительно, аналогов не видно. Четыре в две молекулы – о, это будет Ru(2+) и октаэдр – очень распространённый тип в химии рутения, тысячи комплексов этого структурного типа. Все шесть – это будет Ru(0) и к.ч. 5 – тоже сомнительно, хотя для того, чтобы это понять, нужно ещё представлять химию нульвалентного рутения – вряд ли он сможет удержать молекулу водорода и не захочет отдать её пару электронов, превратившись в комплекс Ru(2+). Ну и вот структура, учтите только, что водороды в ней расположены в значительной степени всё равно гипотетически: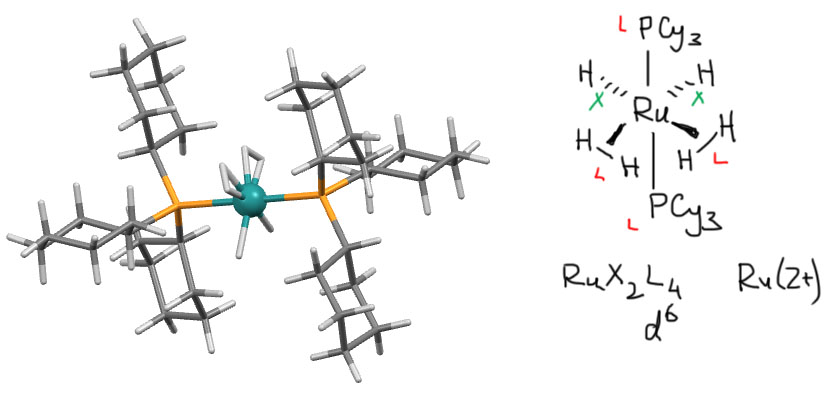
Гапто-лиганд в роли мостика
Гапто-лиганд вполне может быть и би- и даже полидентатным, о чём в следующем разделе, и даже мостиковым. Особенно интересный случай – ацетилен, его тройная связь. Мы отлично знаем, как она устроена – что это две π-связи и они перепендикулярны. То, что их две – это простая идея, и в обычной органической химии мы присоединяем к тройной связи два раза. Но – всегда последовательно, сначала один раз, после чего остаётся обычная двойная связь, потом второй раз. То, что они перпендикулярны, мы в обычной органической химии, в принципе, тоже можем узнать, если изучим сопряжение с разными π-донорами или акцепторами по обеим сторонам тройной связи. Но это как-то немного умозрительно, нужно долго крутить конформации, что-то считать.
А вот с помощью переходных металлов это можно увидеть почти невооружённым глазом, разглядывая картинку структуры комплексов. Каждая из π-связей ацетилена может совершенно отдельно связываться с атомом металла, и в этом случае молекула ацетилена связывает два атома металла, работая как мостиковый лиганд (μ-ацетилен). А в структуре вы прямо и увидите, что атомы металлов расположены перпендикулярно друг к другу – фактически вы прямо видите эти самые перпендикулярные π-орбитали. Таких комплексов довольно много у самых разных поздних переходных металлов, и они играют очень интересные роли в органической химии.
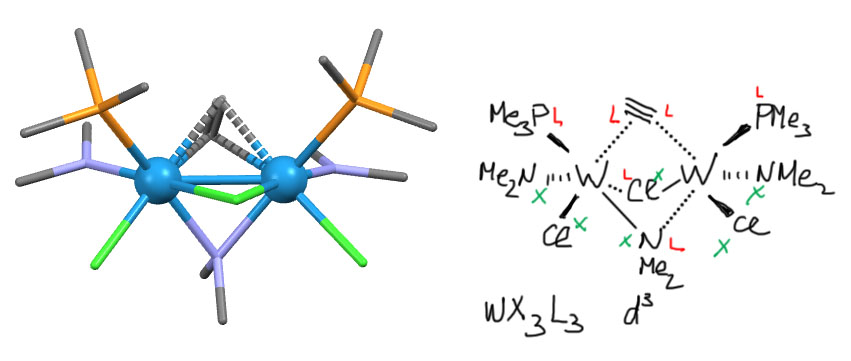
Посмотрим на какой-нибудь комплекс с таким ацетиленовым мостиком. Вот, например, комплекс вольфрама (K.J.Ahmed, M.H.Chisholm, K.Folting, J.C.Huffman, J.Am.Chem.Soc. (1986), 108, 989). Ацетилен в нём, ровно как и предсказано, обслуживает два атома вольфрама на перепендикулярных направлениях. Но комплекс сложный, у элементов из середины блока d-элементов комплексы вообще почти всегда имеют устрашающий вид. Но разобраться можно. Во-первых, на каждом атоме вольфрама мы видим ещё простые лиганды – фосфин, амид и хлорид. Мы легко их классифицируем. И еще один хлорид и один амид, которые работают мостиками. Хитрость в том, что поскольку мостики такого типа сделаны так, что лиганд с одной стороны виден, как X- а с другой как L, мы можем произвольно назначить эти роли. И сделаем это так, чтобы атомы металла имели одинаковую степень окисления. А почему? Почему нельзя отправить оба X-конца на один атом, а оба L-конца – на второй? Комплекс получится с виду точно такой же, но атомы вольфрама будут иметь разную степень окисления – один на 2 единицы больше другого. Возможно ли такое? Вполне, и такие комплексы, в состав которых входят атомы одного металла в разных степенях окисления (смешанно-валентные комплексы) вполне хорошо известны – их много и с успехом изучал нобелевский лауреат Генри Таубе. Один такой комплекс вообще известен всем – это знаменитая берлинская лазурь (или турнбуллева синь – это вообще два комплекса или один? – в том-то и дело, что один). Фокус в таких комплексах в том, что атомы металла просто обмениваются электронами, и если разность степеней окисления равна единице, то такой обмен не сможет уравнять степени окисления, а приведёт к очень интересному явлению, внешним проявлением которого как раз и является глубокий цвет такого комплекса, и за которое, собственно, нобелевку и дали. А если разность степеней окисления два, как у нас здесь, то всё будет очень просто, и один атом отдаст другому один электрон, и атомы станут одинаковыми. Мы даже этого не заметим. Отлично. Маркируем лиганды, считаем степень окисления, получаем W(3+) и конфигурацию d3. Три – это нечётное число (да!), то есть один (минимум) неспаренный электрон на каждом атоме. А когда у соседних атомов есть по неспаренному электрону, логично предположить, что между ними образуется связь. В обычной органической химии мы назвали бы это рекомбинацией радикалов. В химии металлов так лучше не говорить, потому что неспаренные электроны на атомах переходного металла – обычное дело (это или нечётное число валентных электронов, или даже чётное, но в высокоспиновом состоянии), но такие атомы (ионы) просто так не слипаются, обазуя связи между атомами металлов. Такие связи обычно очень слабы, и смысла в них не очень много, … если только такие атомы и так сближены друг с другом, например, теми же мостиками. Тогда связь металл-металл просто становится неизбежной, что, во-первых, видно по реальным расстояниям между атомами в структуре, и во-вторых, экспериментально видно по всяким свойствам, связанным с магнитными моментами неспаренных электронов. Так или иначе, на структуре такая связь изображена палкой между атомами металла. Значит, мы имеем двухядерный комплекс со связью металл-металл – а это настоящий кластер. Этот комплекс – настоящий двухядерный кластер. По-моему, симпатичный…
Не путаем гаптность и хелаты. И строим хелаты из дигаптных центров.
Бидентатный лиганд не нужно путать с дигаптным. Сходство их очень поверхностно – и тот, и другой формально связан с металлом через два атома. Но в хелатном лиганде каждый атом – отдельный лигандный центр, каждый отдает пару, а весь хелат считается как 2L, 2X или LX, в зависимости от конкретной природы координационных центров.
Дигаптный лиганд только формально связан с металлом через два атома: реальный координационнй центр это связь между ними. И обязательно оба атома в дигаптном лиганде должны быть рядом. В среднем примере на предыдущем слайде в комплексе рутения два бидентатных хелатных лиганда, и один дигаптный диводород (да, нужно говорить именно так – диводород!).
Но может быть и такая ситуация, когда хелатный лиганд содержит гаптные координационные центры. Особенно часто это встречается, когда в лиганде несколько изолированных двойных связей – каждая связывается как дигаптный лигандный центр. Несколько таких олефинов просто невероятно популярны в координационной химии: циклооктадиен (сокращенно COD), норборнадиен (NBD), циклододекатриен (CDT)) и множество других.Самый популярный из них COD – бидентатный η2,η2-лиганд – обратите внимание, что он изображается, сложенным в конформацию ванны (в остальном мире это называется лодкой, boat).
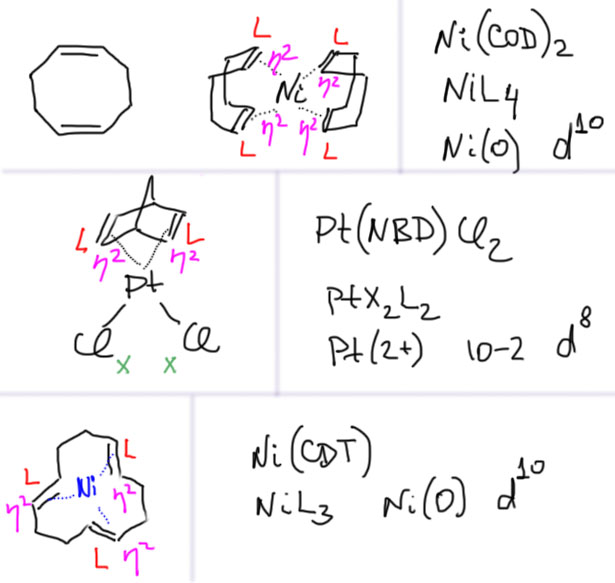
Увеличиваем гаптность…
Возьмем теперь сопряженный диен, хотя бы 1.3-бутадиен, которым нам все уши прожужжали в курсе органической химии. Дает он комплексы? Дает, с удовольствием, только металлы подставляй. Но как их рассматривать, как в предыдущем слайде в виде η2,η2-хелатов с координацией по двум двойным связям? Или бутадиен работает, как один η4-лиганд? Странность в том, что можно и так, и так, но второй способ корректнее описывает реальную структуру, так как бутадиен (точнее, его s-цис-конформер) участвует в связывании с металлом обеими своими связывающими орбиталями, делокализованными по всем четырем атомам углерода сопряженной системы, причем одна из них (нижняя) дает σ-связывание, а вторая (ВЗМО) π-связывание с одной из dπ-орбиталей. Но мы не будем этого рисовать и разбираться подробнее не будем, просто запомним, что сопряженные диены могут работать как единый лиганд η4-типа. Это очень хорошо видно на структурах.
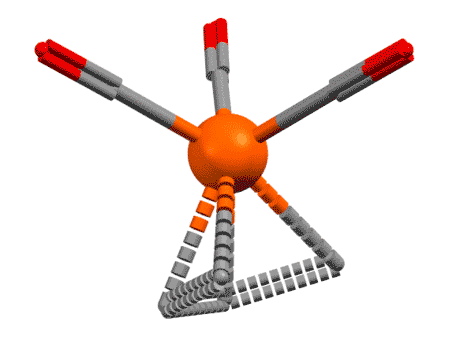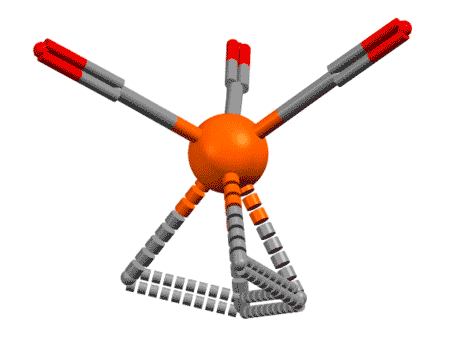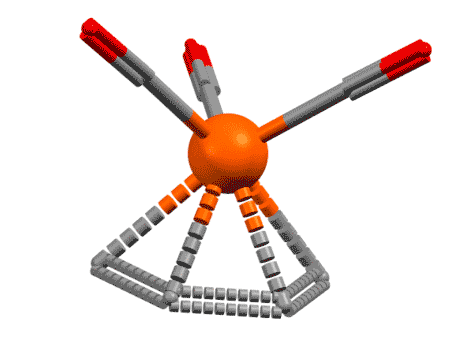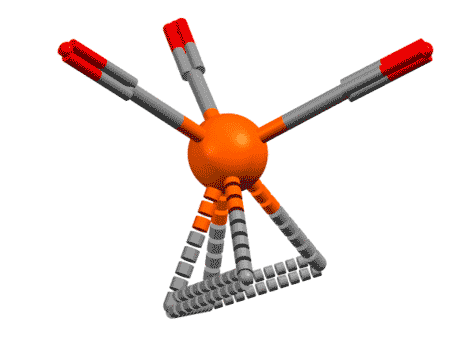
Если бы диен работал как две двойные связи, каждая из них, а следовательно и плоскость диена в целом были бы перпендикулярны оси металл – центр диена. Но в реальности диеновый лиганд наклонён относительно этой оси на довольно значительный угол. Этот наклон связан с тем, что в связывании участвуют орбитали всей сопряжённой системы как целого, и, форма этих орбиталей и определяет максимум взаимодействия с d-орбиталями металла. Металл при этом предоставляет две вакантные орбитали, а лиганд 4 электрона, то есть две пары. Именно поэтому для всех формальных вещей типа подсчёта электронов, координационного числа и т.п. его и можно рассматривать просто как два L-лиганда, и не заморачиваться введением еще одного типа лигандов. И то же самое будет верно для всех подобных лигандов.
Этот комплекс железа очень интересен и с исторической точки зрения. Он мог бы стать тем, чем стал ферроцен, но на четверть века раньше. Этот комплекс получил довольно случайно немецкий химик Ганс Райлен (Reihlen, H.; Gruhl, A.; Hesslein, G. v.; Pfrengle, O. Liebigs Ann.Chem. 1930, 482, 161). Естественно в это время никто не мог понять, как это устроено и зачем это нужно, хотя сам Райлен нарисовал две весьма любопытные структуры.
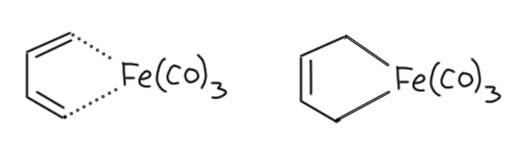
Вторая особенно интересна – такой гетероцикл, как бы мы нынче сказали, феррацикл. Бутадиеновые комплексы бывает такой структуры, мы еще с этим столкнёмся. Но не этот. Установить, как был устроен этот комплекс тогда было нельзя. Описать его так. что на него кто-то обратил бы внимания до ферроцена тоже. Так и остался незамеченным до тех времен, когда с этим разобрались. Сам Райлен был занятным парнем. Судя по этой статье весьма не глупым. Судя по жизни – есть вопросы. Что-то его всё на войну специальные военные операции тянуло. На Первой мировой СВО он оттрубил от первого дня до последнего, правда не пехотинцем, а оператором военного дирижабля – немцы сильно увлекались этими штуками, даже бомбили с них, прямо вручную бомбы кидали. Но и на Вторую мировую СВО, хотя было ему уже под полтинник, он ушёл добровольцем. Причём нигде нет никакихсведений о том, что он сильно увлекался фюрером. Просто такое представление о долге, непонятно только перед кем и чем. Ему повезло, потому что командиры оценили его состояние и решили, что он малопригоден для подвигов, и безжалостно прогнали с передовой. Что и помогло ему дожить до 1950-го. Когда нам иногда приходят в голову вопросы, как может человек добровольно пойти чёрт знает куда, уместно вспомнить этого деятеля и понять, что даже явно очень неглупый профессор университета может иметь в той части головы, которая не отвечает за его науку, что-то типа манной каши.
И ещё…
Лигандом легко может быть и ароматическое соединение, например, бензол, и таких комплексов огромное количество. Комплекс Cr(0) с двумя молекулами бензола, бис(η6-бензол)хром – основополагающая молекула в истории координационной и металлоорганической химии наряду с ферроценом. Впервые полученная выдающимся немецким химиком Эрнстом Отто Фишером в 1955, четыре года спустя после открытия ферроцена, она натолкнула на идею о возможности сендвичевого типа связывания. За установление и доказательство структуры этих двух комплексов Э.О.Фишер и Дж.Уилкинсон получили Нобелевскую премию. Интересно, что для такой важнейшей молекулы как бисбензолхром, сэндвичевый характер установили быстро, а вот с деталями возились почти сорок лет, и очень долго считали, что в бензольных кольцах длины связей неравны и существует немаленькое чередование длин связей, свидетельствующее о не совсем идеальной ароматичности. Это не ошибка, а реальная склонность этой молекулы к такому, как это называется, альтернированию, с очень небольшим барьером активации, что просто приводит к тому, что если условия определения структуры приводят к наблюдению усреднённой конфигурации, наблюдается симметричная форма с осью 6 порядка. А искажение, скорее всего, объясняется просто – нульвалентный хром способен к обратному донорному эффекту, о котором мы поговорим очень скоро – это приводит к частичному переносу электронной плотности на ароматические кольца с металла, что приводит и к появлению зарядов на металле и на кольцах, и к избыточной электронной плотности на ароматических кольцах. Посмотрите когда-нибудь, как выглядят структуры обычных производных бензола с сильными донорными группами (анилинов каких-нибудь и т.п.) – увидите это самое альтернирование и не увидите идеального бензольного кольца. Ароматичность вообще это идеал, достижимый только в нескольких идеальных молекулах типа самого незамещённого бензола, а в большинстве реальных молекул мы видим те или иные отклонения от идеального поведения ароматической системы (альтернирование связей, не очень явно выраженный эффект кольцевого тока, сомнительная стабильность и т.п.). 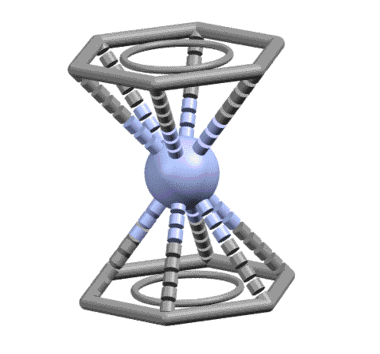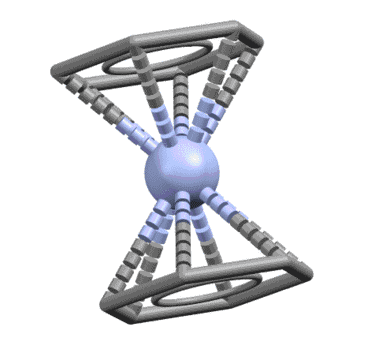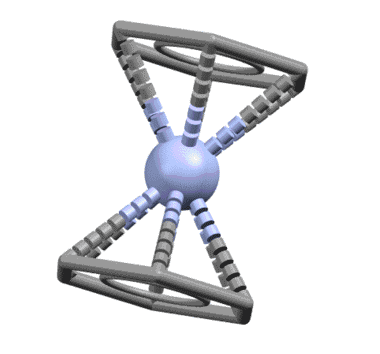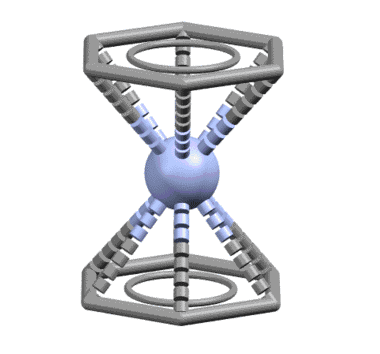
Понятно, что в этом случае из общих соображений – мы же с молоком матери впитали идею о том, что ароматическая система бензола есть незыблемый монолит – должна работать вся сопряженная система, на которой находятся 6 электронов. Чтобы их принять у металла должно найтись три вакантные орбитали. Бензол работает, как один η6-лиганд. Но с формальной точки зрения (6 электронов = 3 пары) бензол отлично представляется как три L-лиганда.
Все вроде бы просто, но, если вдуматься, ситуация довольно неприятная. Мы должны были начать вдумываться уже когда рассматривали бутадиен, но как-то там это в голову не пришло. Но в случае с бензолом на это трудно не обратить внимания. Вот вопрос: связь металла с бензолом – это одна связь или три связи? Если связь одна, то что это за 6-электронная семицентровая связь, мы ведь как-то привыкли к тому, что химические связи обслуживаются парой электронов. Если связи три, то есть металл с бензолом связан тройной связью, то почему мы так не рисуем. Ответ разочарует кого угодно: “Это не важно, как удобно, так и рисуем”. В координационной химии нет такой же железобетонной системы обозначения химических связей и вообще строения молекул, как в органической химии, впрочем, да и там эта строгость кажущаяся – всем известно, например, что нет единого и полностью всех удовлетворяющего способа отображения делокализации, особенно ароматической и антиароматической. Но в органической химии хотя бы простые структуры рисуются однозначно, двойная связь рисуется двумя черточками, тройная тремя. А в координационной химии нет ничего подобного, и это очень часто вызывает ошибки не только в учебной, но даже и в настоящей исследовательской литературе.
В случае сложных лигандов пунктирной линией просто обозначают сам факт связывания металл-лиганд, а природу этой связи или связей рассматривают отдельно, используя молекулярно-орбитальные представления, или иные квантово-химические концепции. В таких формализованных способах рассмотрения можно вообще уйти от разговоров о химической связи, и смотреть на всю структуру в целом. Поэтому, это не так важно, как может показаться. Химия – наука экспериментальная, нам важно, как выглядит молекула, какие у неё свойства и реакционная способность. И любая концепция, которая удовлетворительно и убедительно описывает хотя бы часть этого комплекта, годится (только для этой части, конечно). Строго говоря, мы вполне могли бы рассматривать связь металл – η6-бензол как тройную связь, включающую одну сигма и две пи-связи, почти как в ацетилене, но с другой природой участвующих орбиталей. Но, в отличие от ацетилена, с которым мы можем делать реакции, разрывая эти связи по одной (например, присоединяя одну и две молекулы брома), в случае комплексов металлов с бензолом (или другими ароматическими соединениями, аренами) найти такие реакции будет непросто.
Впрочем, хорошо известно, что бензол и другие арены не обязательно образуют комплексы как η6-лиганды, бывают и η4-связывание (в этом случае из бензольного кольца выделяется диеновая часть), и η2-связывание с одной из двойных связейи такие комплексы играют большое значение в реакциях аренов в координационной сфере металла. Фактически, металл может вытаскивать из целой ароматической системы неароматические части, при этом понятно, что ароматичность в таком кольце накрывается тазом, обычно только не медным, а, например, рутениевым. И для нас это может быть удивительно, ведь ароматичность, особенно в такой молекуле как бензол, это так хорошо, что кто же посмел её нарушить? И мы понимаем, что связь с металлом в таких комплексах – это очень сильная связь, недаром же она фактически тройная. Энергия такой связи вполне компенсирует потери при нарушении ароматичности.
Способ связывания зависит от многих факторов. Безусловно, в большинстве случаев η6-связывание, сохраняющее ароматичность бензола, будет преобладать. Но может сложиться такая ситуация, что металл видит бензол и хочет укусить его за все шесть электронов, а нечем, валентных возможностей не хватает. Что такое эти валентные возможности, и как их можно прикинуть, мы очень скоро разберёмся. А пока посмотрим на очень характеный пример – два комплекса рутения с гексаметилбензолом одинакового состава, один в степени окисления +2, другой в нульвалентном состоянии. И если первый комплекс (M.D.Ward, D.C.Johnson, Inorg.Chem. (1987), 26, 4213) совершенно симметричен и очень похож на самый типичный комплекс такого типа, бис(бензол)хром, то комплекс нульвалентного рутения (G.Huttner, S.Lange, Acta Crystallogr.,Sect.B, 1972, 28, 2049) не может взять второе кольцо целиком, и становится несимметричным. 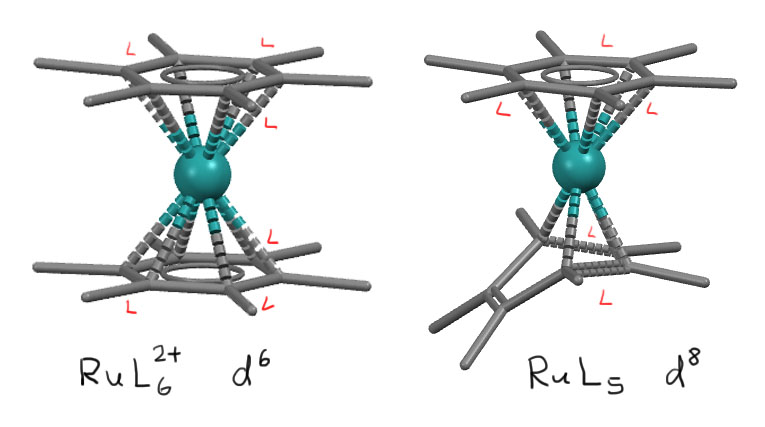
Аллильная система
Из органической химии мы хорошо знаем, что такое аллил, и как одновалентный остаток, и как делокализованная система (аллильный катион, радикал, анион). Если мы попробуем представить себе, как устроен аллил как лиганд, то первое, что приходит в голову, это – простой лиганд X-типа, похожий на метил, этил, пропил и т.п. Да, таких комплексов довольно много. Но в такой системе есть и двойная связь, и почему бы металлу, если у него еще есть вакантная орбиталь, не подхватить эту связь как дигапто-лиганд L-типа. Но как только мы это представим, получим, что в связь с металлом вовлечены три атома углерода подряд, то есть получится η3-лиганд с формальным типом связывания X+L. Нужно очень точно понимать, что такое этот формальный тип связывания. Это именно формальный, абстрактный приём, облегчающий учёт лиганда в координационной сфере металла. Он не имеет никакого отношения к реальному связыванию лиганда с металлом, с природой связей, молекулярными орбиталями и всему тому, что даёт нам понимание, как устроен тот или иной комплекс. В реальном η3-аллильном лиганде нет никаких отдельных X и L центров, точно так же, как в η6-бензоле нет трёх разных L-центров. Приём формального разделения целого гаптного лиганда на простые типы связывания работает только для того, чтобы быстро прикинуть количество электронов, общий заряд и другие формальные параметры комплексообразования.
В реальности, аллильные комплексы, которые невероятно широко распространены и очень популярны, как правило имеют η3-строение, но в растворах очень легко обратимо изомеризуются в простой аллильный тип и обратно, с потерей первоначального места связывания. Из-за этой изомеризации, с такими комплексами часто бывает невозможно сделать селективные реакции, если не позаботиться о других лигандах, которые будут это безобразие контролировать. Мы к этому еще вернемся.
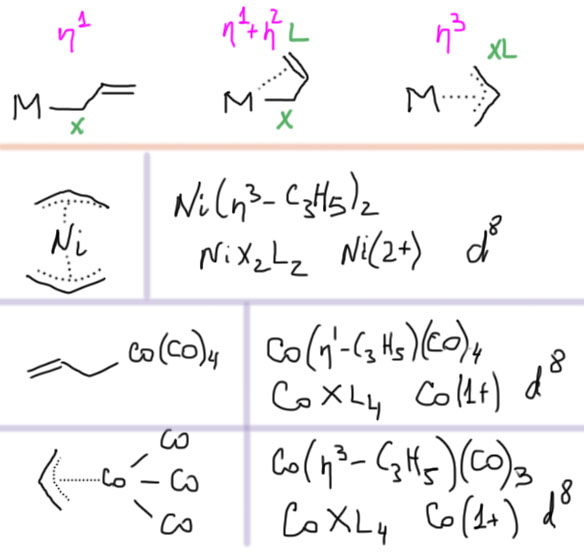
На структурах хорошо видно, что аллильная система действительно связывается с металлом целиком, и если аллил незамещённый или замещённый симметричный, аллильный фрагмент полностью симметричен с одинаковыми расстояниями крайних атомов до металла. Атомы водорода или заместители чуть-чуть отклоняются от плоскости в сторону от металла.
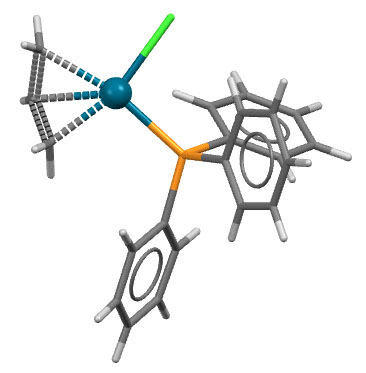
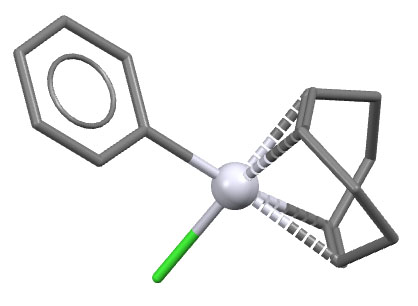
Структура комплекса PdCl(η3-аллил)PPh3 (S.P.Gubin, L.G.Kuzmina, L.A.Polyakova, A.V.Churakov, ccdc 101525). Видно, что это типичный комплекс d8 с плоско-квадратной конфигурацией.
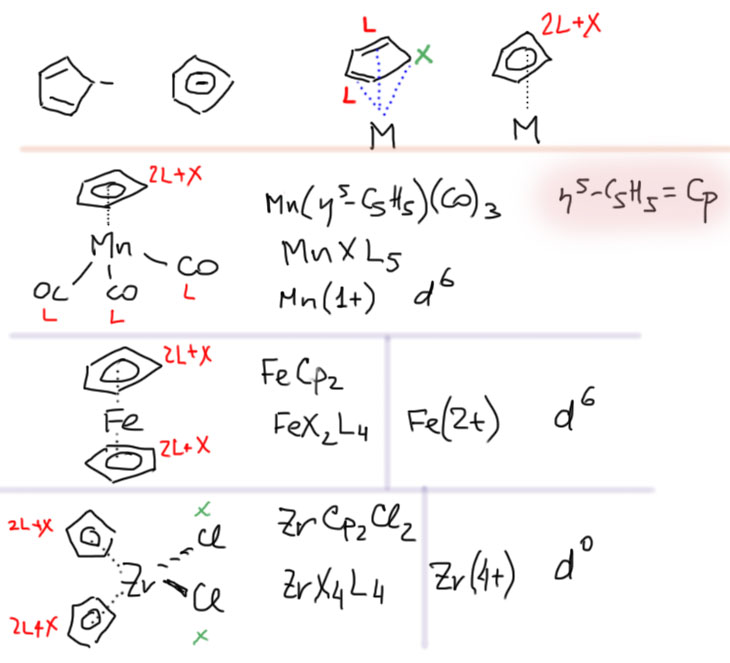
Циклопентадиенильные лиганды
После аллильной системы уже не составит труда разобраться со священной коровой координационной химии переходных металлов – циклопентадиенильной системой. С открытия ферроцена все это и начиналось. Циклопентадиенильных комплексов, без преувеличения, миллион, и нет такого металла, который бы не имел таких производных.
Циклопентадиенил в виде аниона – ароматическая система с 6 электронами, и в этом смысле она похожа на бензол. Если рассмотреть ее как лиганд, то можно сделать то же самое, что мы делали с аллильной системой – сначала подцепить металл на одну валентность в виде X-лиганда, а затем связать две двойные связи как два L-лиганда. Так как все эти координационные центры рядом, то полная картина связывания соответствует η5-гаптности, но в отличие от бензола здесь формальный 2L+X тип связывания, и каждый Cp-лиганд (это общепринятое сокращение пентагапто-циклопентадиенильного лиганда) дает +1 в степень окисления металла (бензол давал ноль). Так же как и для аллильного лиганда подчеркиваю, что это разбиение целого лиганда на 2L+X совершенно формально, и служит только для удобного и быстрого подсчёта формальных параметров комплекса, но совсем ничего не говорит о его реальной структуре. Собственно, мы с тем же успехом могли бы учитывать циклопентадиенильный лиганд как 3L-лиганд с зарядом -1. Получилось бы то же самое, только нам пришлось бы не забывать дать металлу заряд +1 на каждый Cp, и учитывать этот заряд в определении степени окисления и числа d-электронов. Это будет немного более громоздкая и менее удобная схема, но при этом и более универсальная. Такая схема не подвела бы нас при работе с ещё более хитрыми комплексами и лигандами, например, тропилиевым (циклогептатриенильным) лигандом, который сейчас невероятно популярен в координационной и органической химии. Пока оставим это, но обязательно когда-нибудь разберёмся.
Все остальное совершенно так же, как всегда – определяем все лиганды, размечаем L/X, находим степень окисления, из нее число d-электронов, и дело в шляпе. Бывают ли, как у бензола, другие способы связывания циклопентадиенильных лигандов? Бывают, но очень-очень редко, это экзотика за пределами этого курса. Не будем об этом думать, для нас Cp и все его производные всегда будут пентагапто. А как там насчет связей, сколько их, какие они? Все почти как у бензола, не будем на этом останавливаться еще раз.
Комплексов с Cp и его родственниками невероятное количество. Запрос в Кембриджскую базу даёт около 46 тысяч структур – это только те, для которых есть надёжный рентгеноструктурный анализ. Можно смело считать, что реально их описано минимум ещё в 10 раз больше – не менее полмиллиона. Вот какую мощь копнули всего немногим более полувека назад Кили и Посон, Отто Фишер и Уилкинсон. Посмотрим на структуры нескольких знаменитых представителей: самого ферроцена – полного сэндвича, марганцевого полусэндвича (это ещё образно называют piano stool complex, комплекс, смахивающий на рояльный стульчик) цимантрена, и косого сэндвича с дополнительными лигандами цирконоцендихлорида.
Циклопентадиенильные лиганды, как правило, бывают пента-гапто, но, как и для бензола известны и другие способы связывания. Довольно часто встречается циклопентадиенил как простой лиганд X-типа, связанный с металлом одной ковалентной связью. Такой способ любят, например, металлы 11 группы, особенно самый большой, золото. Собственно, понятно почему. У этих металлов своих электронов очень много, и если добавить еще и пента-гапто-Cp, то уже будет 16 электронов, и место только ещё для одного L-лиганда. Такие комплексы действительно есть у меди и серебра, а вот золото предпочитает ухватиться за Cp через один углерод. У такого комплекса есть понятная особенность. Если ухватить циклопентадиенил за один атом, мы получим нечто, очень похожее на аллильный комплекс с η1-связыванием. Но когда мы рассматривали аллильные комплексы, мы видели, что у них обычно бывает равновесие между η1– и η3-связыванием, которое усредняет положение лиганда между первым и третьим углеродом. А когда мы то же самое применим к η1-связыванию циклопентадиенила, мы поймём, что таким способом металл будет обходить все пять углеродов цикла. Иными словами, в моногапто-циклопентадиениле металл всё равно свяжется со всеми пятью атомами цикла, только не одновременно, а последовательно, и этот процесс можно заморозить, и при низкой температуре в рентгеноструктурном анализе получить ожидаемую структуру. Вот как у комплекса золота с η1-Cp и триизопропилфосфином (H.Werner, H.Otto, T.Ngo-Khac, C.Burschka, J.Organomet.Chem. 1984, 262, 123). 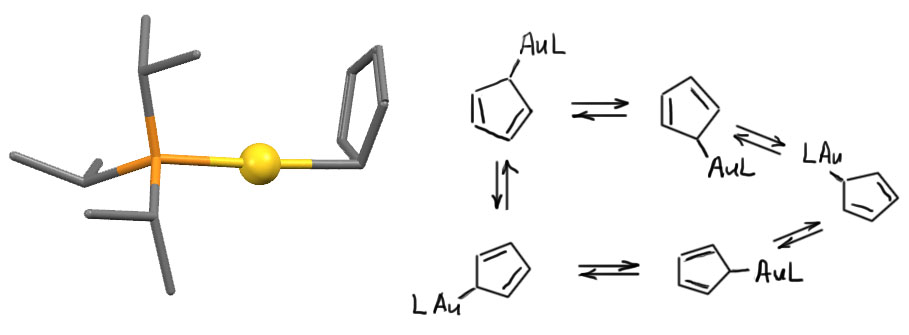
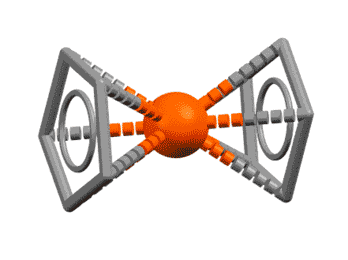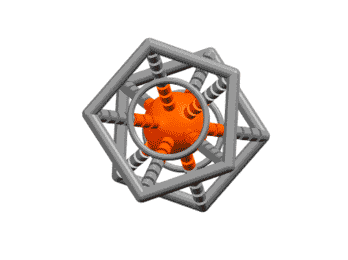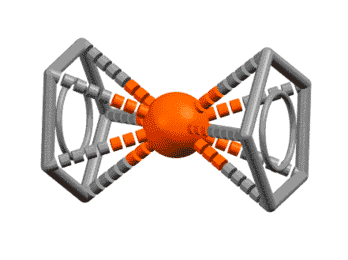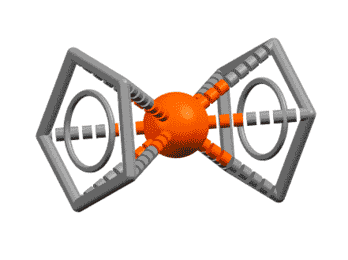
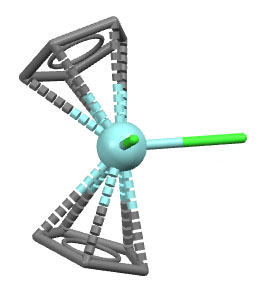
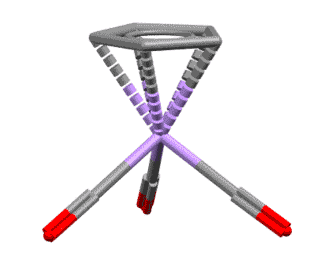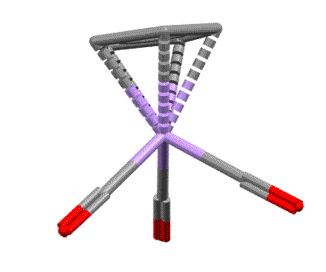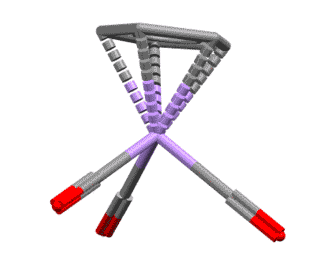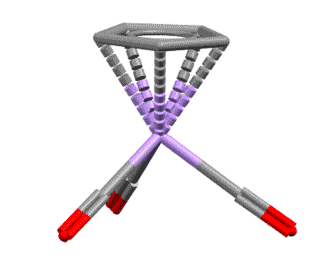
Вот несколько примеров комплексов, в которых есть лиганды с одной, двумя, тремя двойными связями, с различными способами связывания. Разберитесь в структурах комплексов, посчитайте электроны, и прочие характеристики комплексов. За полную характеристику, включая счёт валентных электронов, до 5 плюсиков за структуру.
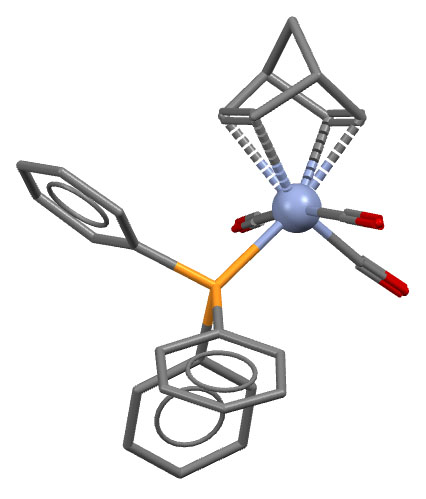
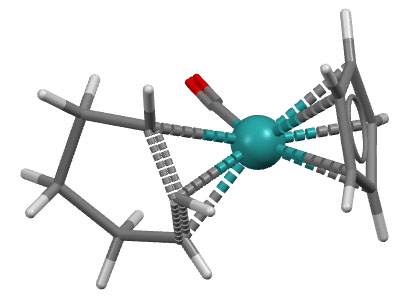
QOVTUR: M.Fianchini, T.R.Cundari, N.J.DeYonker, H.V.R.Dias, Dalton Trans. 2009, 2085, doi:10.1039/b902678a
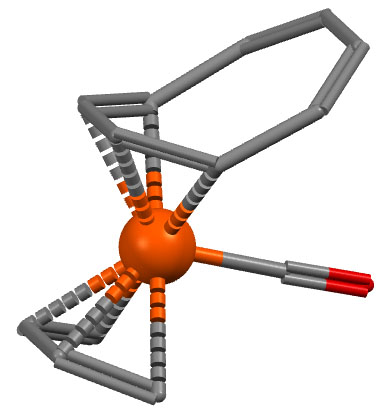
YASPOX01: A.L.Rheingold, D.S.Glueck, C.Scriban, CSD Communication 2005
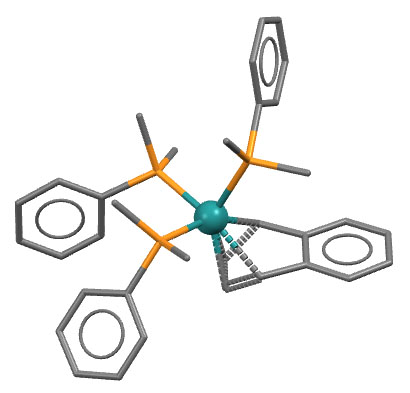
QUDWES: A.K.Renfrew, A.D.Phillips, E.Tapavicza, R.Scopelliti, U.Rothlisberger, P.J.Dyson, Organometallics 2009, 28, 5061, doi:10.1021/om900345n
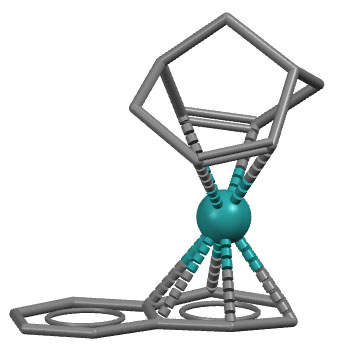
NBCRCP: J.P.Declercq, G.Germain, M. van Meerssche, S.A.Chawdhury, Acta Crystallogr.,Sect.B:Struct.Crystallogr.Cryst.Chem. 1975, 31, 2896, doi:10.1107/S056774087500917X
BUTDMO11: M.Kaupp, T.Kopf, A.Murso, D.Stalke, C.Strohmann, J.R.Hanks, F.G.N.Cloke, P.B.Hitchcock, Organometallics 2002, 21, 5021, doi:10.1021/om020525v
DAZFOZ: M.Crocker, M.Green, C.E.Morton, K.R.Nagle, A.G.Orpen, J.Chem.Soc.,Dalton Trans. 1985, 2145, doi:10.1039/dt9850002145
Вот несколько примеров комплексов, в которых есть полигаптолиганды ароматического или неароматического типа. Разберитесь в структурах комплексов, посчитайте электроны, и прочие характеристики комплексов. За полную характеристику, включая счёт валентных электронов, до 5 плюсиков за структуру.
Комплекс железа с бутадиеном, циклооктатетраеном и CO.
I.W.Bassi, R.Scordamaglia, J.Organomet.Chem. 1972, 37, 353
Комплекс рутения с нафталином и диметилфенилфосфином.
M.A.Bennett, M.Brown, D.C.R.Hockless, Aust.J.Chem. 2000, 53, 507
Комплекс рутения с нафталином и бицикло[3.3.1]нона-2,6-диеном.
Y.Hiroi, N.Komine, S.Komiya, M.Hirano, Org.Lett. 2013, 15, 2486
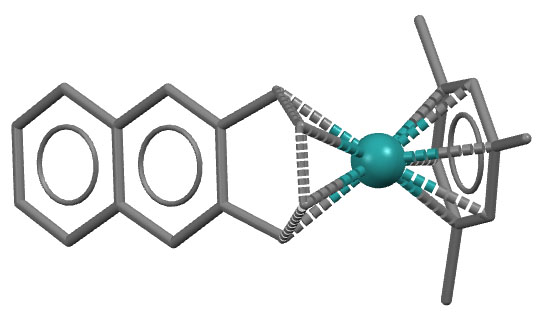
Комплекс рутения с антраценом и 1,3,5-триметилбензолом (мезитиленом)
А.С.Бацанов и др., Металлоорганическая химия, 1988, 1, 326
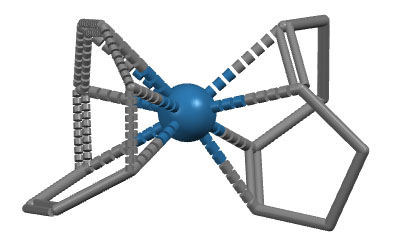
Комплекс осмия с 1,5-циклооктадиеном и циклооктатетраеном: Os(COD)(COT)
Jia En Low, A.Foelske-Schmitz, F.Krumeich, M.Worle, D.Baudouin, F.Rascon, C.Coperet, Dalton Trans., 2013, 42, 12620
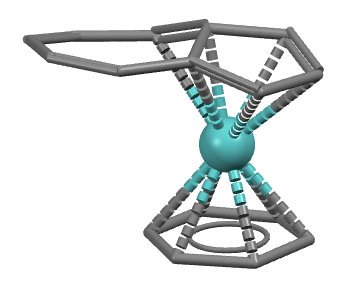
Комплекс молибдена с азуленом и бензолом
S.Tofke, U.Behrens, Angew.Chem., Int.Ed., 1987, 26, 147