Декарбонилирующие реакции
Так как миграционное внедрение всегда реакция обратимая, можно придумать реакции, в которых ацильный комплекс металла получается каким-то другим способом, не из миграционного внедрения в карбонильном комплексе. Очевидный кандидат – окислительное присоединение к каким-нибудь производным карбоновых кислот. Ацильный комплекс имеет все шансы превратиться в карбонильный обратной реакцией к миграционному внедрению. Это выглядит так, как будто переходный металл вырывает карбонильную группу из производного карбоновой кислоты. И если на 3 курсе вам показалось неубедительным, что карбоновые кислоты и их производные называют карбонильными соединениями наряду с альдегидами и кетонами, вот отличное доказательство – так считают переходные металлы, а с переходными металлами спорить бесполезно. 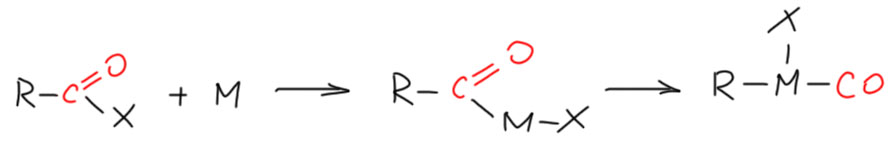
Почему идут такие реакции? Обычно потому что карбонильные комплексы переходных металлов очень стабильны, ведь CO – очень хороший лиганд, умеющий пользоваться всеми валентными возможностями переходного металла. Скорее, направить реакцию карбонильного комплекса в сторону σ-ацильного комплекса сложно – это делают далеко не все переходные металлы, а кроме того всегда нужна помощь анциллярных лигандов. А наоборот – нет проблем. Некоторые переходные металлы, например, родий и рутений так любят карбонильный лиганд, что вырывают карбонил из альдегидов, а иногда даже из спиртов.
Но в этом месте ограничимся производными карбоновых кислот. Чтоб мы вообще хотим получить? Пока не знаем, просто хотим попробовать, ведь нам для начала непонятно, какой переходный металл может захотеть окислительно присоединиться к производному карбоновой кислоты. Но из общих соображений можно подумать, что производное нужно взять наиболее активное – галогенангидрид. Хлорангидрид. Окислительное присоединение, как мы уже знаем, предпочитает более тяжёлые галогены, но в галогенангидридах хлорангидрид оказался вполне реакционноспособным. То, что хлорангидриды неплохо реагируют в реакции Мидзороки-Хека и Стилле, обнаружили очень рано – собственно в случае кросс-сочетания уже первые работы и Стилле и Мигиты-Косуги использовали ацил хлориды наряду с обычными субстратами и даже раньше их (M. Kosugi, Y. Shimizu, T. Migita Chem. Lett., 1977, 1423; D. Milstein, J. K. Stille J. Am. Chem. Soc. 1978, 100, 11, 3636; ). Но – и у тех и у других получились … кетоны. 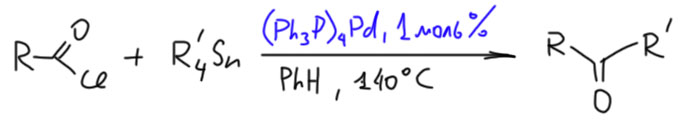
То есть это обычное кросс-сочетание, только в качестве электрофила используется ацилхлорид. Но когда несколько лет спустя Спенсер попробовал ацилхлорид в реакции Мидзороки-Хека получился не кетон, а произошло декарбонилирование (H.-U.Blaser, A.Spencer J. Organomet. Chem., 1982, 233, 267). 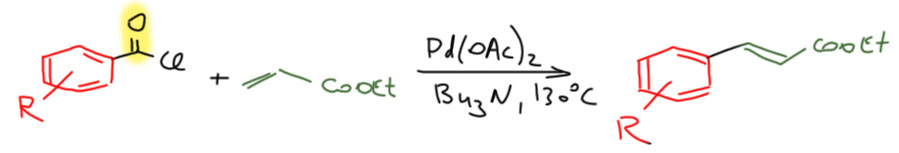
В дальнейшем, хлорангидриды применения в качестве субстратов не получили.
Вопрос: разница между кросс-сочетанием и реакцией Мидзороки-Хека
Результаты с использованием хлорангидридов в кросс-сочетании и реакции Мидзороки-Хека ярко иллюстрируют принципиальную разницу между этими типами реакций. Напомню, что реакция с оловоорганикой даёт кетоны и идёт без декарбонилирования, а реакция с олефином идёт с ацетатом палладия без фосфина и даёт продукт декарбонилирующего Хека. Более того, добавка фосфинового лиганда полсностью блокирует реакцию – она просто не идёт.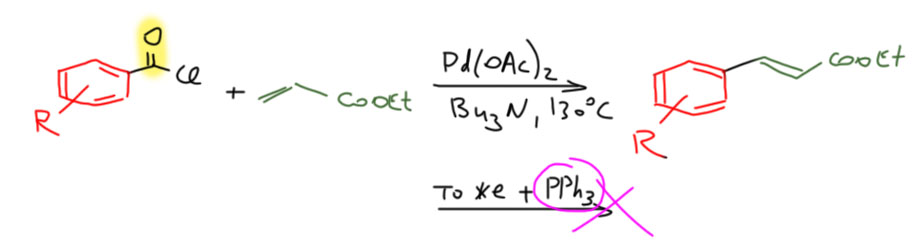
Попробуйте нарисовать каталитический цикл декарбонилирующего Хека и объяснить, почему он блокируется фосфиновым лигандом. И почему в кросс-сочетании ничего подобного нет, и фосфин реакцию не блокирует, но получается кетон.
Реакция Мидзороки-Хека без оснований – подмена лиганда
После хлорангидридов по реакционной способности следуют ангидриды. Но то, что работает в обычной органической химии, не всегда справедливо в химии переходных металлов. Окислительное присоединение к связям C-O идёт намного хуже, чем к связям C-галоген (хотя мы знаем исключение в образовании комплексов η3-аллильного типа). Поэтому неудивительно, что даже до ангидридов дело дошло нескоро. Но в 1998 голландский исследователь Йоханнес де Фрис, большой энтузиаст бесфосфинового катализа и палладиевых наночастиц, с сотр. описал занятную модификацию реакции Мидзороки-Хека. В эту реакцию вступают ангидриды с бесфосфиновым катализатором, при весьма высокой температуре в полярном высококипящем растворителе (NMP – это N-метилпирролидон, один из аналогов ДМФА, более дешёвый и устойчивый при высоких температурах реакции, за что его очень любят в крупномасштабных, промышленных и полупромышленных, применениях). Интереснейшей особенностью реакции было отсутствие оснований, необходимых для регенерации Pd(0) в каталитическом цикле Хека. Но требовалась небольшая добавка бромида натрия, всего 1 моль%, чуть-чуть больше, чем загрузка самого ацетата палладия. 
Как же регенерируется катализатор? Бромид вряд ли может работать как основание.
Это очередной пример того, как переходный металл выбирает себе лиганды. Палладий предпочитает галогениды кислородным нуклеофилам. Нарисуем каталитический цикл. В нём будет больше обычного стадий, и порядок некоторых из них в реальности не обязательно такой, как показано (например, когда точно уходит CO и когда точно происходит превращение ацила в карбонил), но это совершенно не важно и вряд ли вообще когда либо будет установлено. В химии вообще всегда приходится иметь дело не с полным, а с частичным знанием, и извлекать из отрывков полезные выводы.
Итак, ангидрид окислительно присоединяется к Pd(0) (это типичный случай бесфосфинового катализа, в котором электрофил перехватывает малоустойчивые частицы Pd(0), не имеющие достаточной лигандной поддержки, а возможный избыток Pd(0) резервируется в наночастицах, играющих роль resting state). Ацил превращается в карбонильный комплекс. Дальше всё могло бы пойти как в обычном Хеке, но в отсутствие основания нечем было бы регенерировать Pd(0) из гидрида, и цикл встал бы. Но помогает бромид – он вытесняет из координационной сферы палладия слабее связанный ацетат, и ацетат вступает в дело как раз как основание, забирающее протон. Основности ацетата для этого достаточно – это хорошо известно из обычной реакции Мидзороки-Хека, в которой ацетат очень любим как основание. Бромид при этом регенерируется, а следовательно нужен только в каталитических количествах. В протоколе де Фриса используется 0.25 моль% палладия и 1 моль% бромида – всего в 4 раза больше, очевидно для того, чтобы сдвинуть равновесие обмена ацетат-бромид.
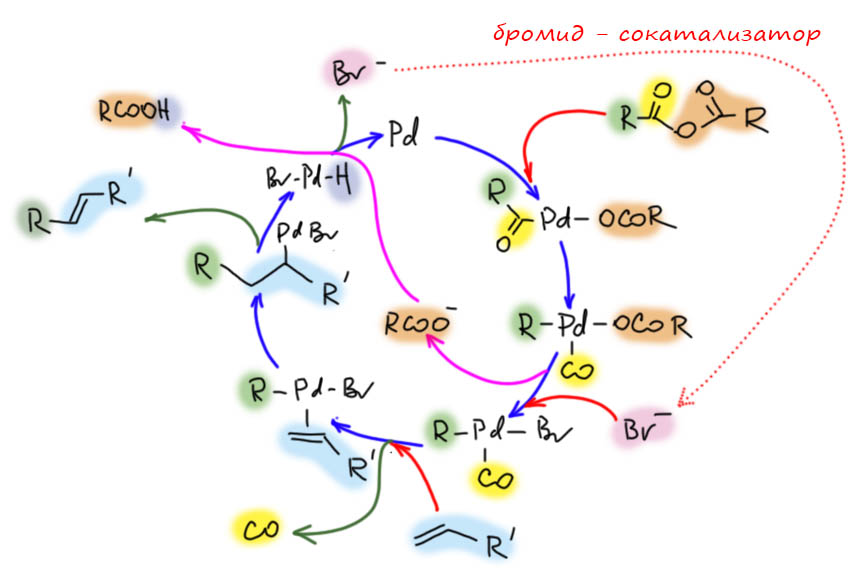
Вот такой цикл, показавший эффективность манипуляций с лигандами. В дальнейших исследованиях каталитических реакций с участием палладия и не только этот приём стали применять очень часто, подсовывая переходному металлу нужный лиганд, вместо того, который образовался на ранних стадиях каталитического цикла.
Реакции сложных эфиров и амидов
Сложные эфиры и, тем более, амиды обладают низкой реакционной способностью как ацилирующие агенты даже в обычной органической химии, а уж для переходных металлов они долго казались совсем безнадёжно бесполезными. Но достижения в понимании роли лигандов и появление новых поколений лигандов сделали то, что так долго и безуспешно ждали советские люди – сказку былью. Появились эффективные протоколы использования сложных эфиров и даже амидов в декарбонилирующих реакциях кросс-сочетания. Другое дело – реакция Мидзороки-Хека. Мы ещё раз убеждаемся в особенности этой реакции – она плохо управляется лигандами, потому что не остаётся места в координационной сфере для размещения серьёзных анциллярных лигандов. Поэтому успехи здесь скорее курьёзны и вполне иллюстрируют пословицу и поговорку “Голь на выдумки хитра”. Голь – это бесфосфиновый палладий. И хитрость заключается в подборе особых субстратов. Немецкий исследователь Лукас Гоосен, большой любитель всяких трюков в катализе переходными металлами, например, взял п-нитрофениловые эфиры (L. J. Gooßen, J. Paetzold, Angew. Chem., Int. Ed., 2002, 41, 1237). Нитрофеноляты – намного лучшие уходящие группы, чем просто феноляты и, тем более, алкоксиды. Их часто применяют в качестве активирующих групп в химии карбоновых кислот в обычной органической химии. Вот и в Хеке они сработали вполне прилично. Условия очень похожи на условия для ангидридов, только ещё пришлось добавить немало изохинолина, который работает как слабый лиганд, немного задерживая деактивацию Pd(0) – рост наночастиц и слипание в чернь. Такие странные и немного беспорядочные приёмы часто применяют в бесфосфиновом катализе. Главная проблема таких приёмов – их столько же, сколько и самих реакций и никогда нельзя сказать, какой сработает.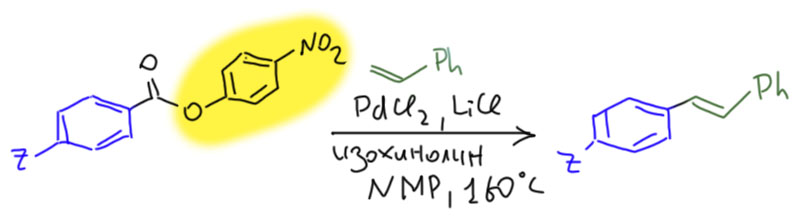
А вот в кросс-сочетании этих ограничений нет, и любителям вкусить радость эксперимента и попробовать раскачать на Судзуки-Мияуру что-нибудь экзотическое – полное раздолье. Надо понимать, что дело это всё равно бешено сложное, – реакционная способность связи C-O очень мала, и окислительно к ней присоединиться не горит желанием практически никто. Но мы знаем, что когда есть проблема с окислительным присоединением, нужно обращаться не к палладию, а к никелю, с его совершенно невероятной реакционной способностью. Но и с этим пришлось повозиться. Первый серьёзный протокол использования сложных эфиров в реакции Судзуки-Мияуры разработали исследователи сразу из трёх лабораторий в Японии и США, Ямагути, Мусаева и Итами, а результат оказался настолько значительным, что его опубликовали в Nature Communications, топовом журнале даже не по химии, а по всем естественным наукам (K. Muto, J. Yamaguchi, D. G. Musaev, K. Itami, Nat.Commun., 2015, 6, 7508). То есть это буквально бомба, а не результат. Видим, что все равно пришлось использовать фениловые эфиры: фенолят лучше как уходящая группа, чем обычные в сложных эфирах алкоксиды. И если вам кажется, что между нитрофенолятом и фенолятом большой разницы не, вы неправы – разница огромна (самые проницательные из читателей могут обнаружить здесь довольно мерзкую недомолвку, но если обнаружите, не расстраивайтесь, в химии полно таких гадостей, и если вы их видите, значит, по крайней мере, вы сами в химии кое-что смыслите, и это прекрасно). Видим, что пришлось не только взять никель, но и дать ему сильнодонорный фосфиновый лиганд. Лиганд выглядит не так модно, как патентованные и брендированные лиганды Бачуолда и Хартвига, но это триалкилфосфин, сильный σ-донор, совсем не обладающий π-кислотностью. 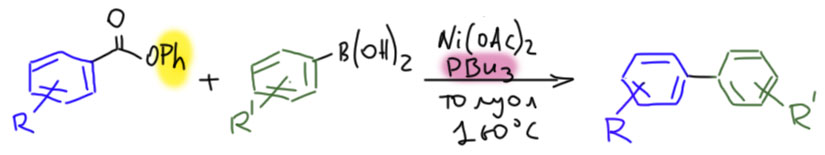
За прошедшие после выхода этой статьи 4 года появилось ещё несколько протоколов, в основном тоже использующие никелевый катализатор с сильнодонорными фосфиновыми лигандами. В одной из этих статей приводятся доводы в пользу того, что окислительное присоединение никеля происходит не по связи C-O, а по связи С-С между ароматическим кольцом и карбонилом (J. Am. Chem. Soc. 2018 140 (10), 3724-3735). Нам сейчас это точно не очень важно, а желающие могут попробовать разобраться детальнее.
С амидами дело еще сложнее. Связь C-N в амидах необычайно прочна, она обладает высокой степенью двоесвязанности из-за сопряжения пары на азоте и карбонила. Влезть в такую связь очень трудно, может быть даже невозможно. Но хочется. Ничто так не возбуждает порядочного химика, как возможность влезть в такую связь, в которую до него никто ещё не влезал. Зачем? Ради спортивного интереса, славы, статьи в гламурном журнале с огромным импактом, и – ради расширения возможностей химии. Никто заранее не знает, что может понадобиться в каком-нибудь будущем исследовании – лучше иметь возможности, чем не иметь их. С амидами впрочем всё настолько тяжко, что потребовалось изобретать специальные субстраты, чтобы хотя бы продемонстрировать принцип. Особенно преуспел в этой химии Шостак с сотр. из США (C.Liu, M.Szostak, Org. Biomol. Chem., 2018, 16, 7998). Он точно сформулировал ключ к решению – нужно задавить сопряжение и другим сопряжением и стерикой, выводящей азот из плоскости. Амиды, которые отвечают этим требованиям выглядят странно, но придётся смириться – других амидов для вас у Шостака и других исследователей, занимающихся вовлечением амидов в декарбонилирующее кросс-сочетание и даже в Мидзороки-Хека нет. Чаще всего используют такое производное, где на азоте целых три карбонила – N-ацилглутаримиды. Вот условия реакции Хека. Основания не требуется. Не правда ли, что-то похожее мы недавно видели – бесфосфиновый катализ, высокая температура, добавка бромида.
А вот кросс-сочетание. Берём никель и даём ему сильнодонорный триалкилфосфин. Условия всё равно очень жёсткие.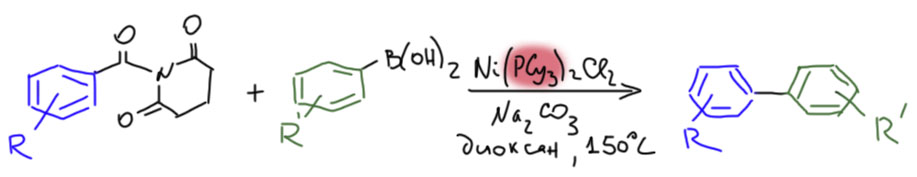
Другие тоже не отставали. Например, китайский исследователь Ши с сотрудниками сделали борилирование по Мияуре (Z. Shi et al, Angew. Chem.,Int. Ed., 2016, 55, 8718), взяв немного другой амид, но тоже имеющий Boc-группу на азоте, ослабляющую C-N связь, опять никель и NHC-лиганд (ICy – догадайтесь сами, что это такое). На боре висит не привычный нам пинакон, а другой очень популярный гликоль для эфиров борной кислоты с тривиальным названием неопентилгликоль, сокращённо nep).
В литературе последних трёх-четырёх лет можно найти ещё несколько занятных реакций, использующих сложные эфиры и специфические амиды для реакций кросс-сочетания, катализируемых комплексами никеля, палладия и родия.
Пример. Декарбонилирующее и недекарбонилирующее кросс-сочетание
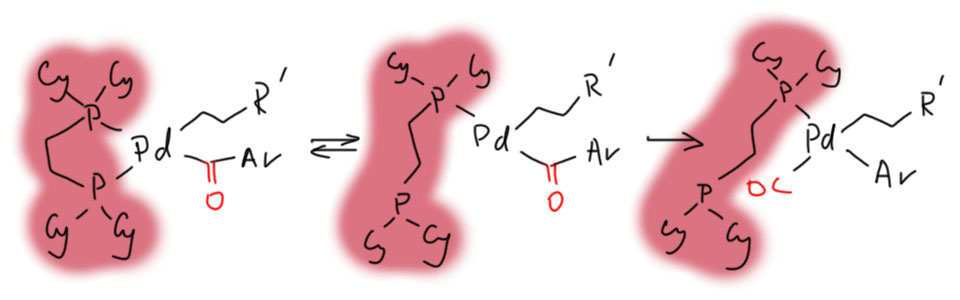
Современная химия научилась намного эффективнее управлять каталитическими процессами с помощью подбора анциллярных лигандов. Интересный пример описан в недавней работе Ньюмена (S.G.Newman et al.2018 20 (13), 4094-4098). Кросс-сочетание продуктов гидроборирования алкенов с сложными эфирами требует сильнодонорных лигандов, и может быть направлено по одному или другому пути. Катализатор с NHC-лигандом (обратите внимание, что использован пред-катализатор аллильного типа, которые мы обсуждали в теме Аллильное замещение) направляет кросс-сочетание к кетону, блокируя декарбонилирование. А хелатный сильнодонорный дифосфин (это аналог dppe, но с циклогексилами вместо фенилов) вроде бы тоже должен хорошо держать координационную сферу, но нет – с ним идёт декарбонилирование.
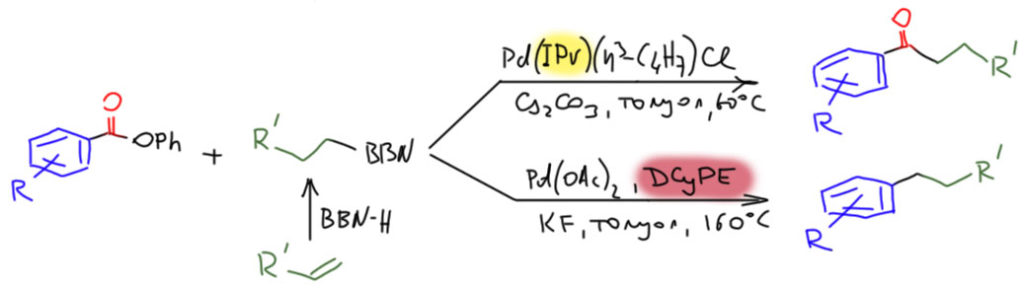
Интересно, что из общих соображений всё должно быть наоборот – монодентатный лиганд должен способствовать декарбонилированию, а бидентатный – блокировать его. Но вмешиваются дополнительные факторы. Посмотрим на каталитический цикл, показав палладий без анциллярных лигандов, только с лигандами-акторами. Грузим на палладий электрофил (сложный эфир), затем нуклеофил (боран с помощью фторида – что делает борид в переметаллировании боранов можно вспомнить здесь). Дальше цикл приезжает к развилке – выбрасывать карбонил или нет. Если выбрасывать – ему нужно доступное координационное место. И в зависимости от выбора можно получить два продукта, с карбонилом и без. И этот выбор определяет анциллярный лиганд. 
Огромный NHC-лиганд, хоть и монодентатный, зато страшный и рогатый, не даёт карбонилу перебраться на формально свободное координационное место и так блокирует декарбонилирование. Реакция даёт кетон.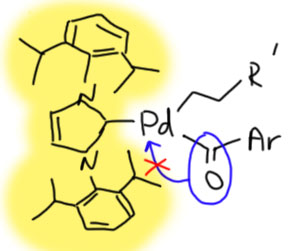
А бидентатный лиганд оказывается не стабильным, а геми-лабильным (геми – это то же самое что полу-), чему способствует и высокая температура реакции – 160ºС, при этой температуре хелатный цикл раскрывается, и место освобождается, потому что вторая фосфиновая группа свободно болтается на гибком поводке. СО пристроить удаётся и получается продукт декарбонилирующей Судзуки-Мияуры.