Гомогенное гидрирование
Гидрирование – это одна из первых каталитических реакций с участием комплексов переходных металлов, которая вошла в органический синтез. Способность переходных металлов и их производных катализировать реакции гидрирования кратных связей хорошо известна из гетерогенного гидрирования. Все знают катализаторы гетерогенного гидрирования: палладий на угле, диоксид платины, никель Ренея и множество более специализированных форм. Гетерогенное гидрирование известно с 19 века, и даже удостоилось своей нобелевской премии в лице французского химика Поля Сабатье в 1912 году за открытие и широкое внедрение высокоактивных мелкодисперсных форм металлов, в частности, никеля. В принципе, для целей гидрирование алкенов и алкинов гетерогенное гидрирование удовлетворяет всем потребностям и лабораторного и особенно промышленного синтеза: катализаторов много на все вкусы и потребности, использовать их очень удобно, так как они не смешиваются с продуктами реакции, и легко отделяются. Особенно это приятно в промышленности, где катализатор может быть использован в поточном режиме, закреплен в реакторе, в который с одной стороны подаются исходные, а с другой выгружаются продукты.
Но у гетерогенного гидрирования все же есть свои недостатки. Самый важный из них – селективность. Гетерогенное гидрирование еще можно научить различать двойные и тройные связи, и мы активно используем эту возможность в селективном гидрировании тройных связей, но вот с выбором разных двойных связей для гидрирования дело обстоит намного хуже. И совсем плохо у гетерогенного гидрирования с асимметрическим, энантио- или диастереоселективным гидрированием. Попыток разработать гетерогенный катализатор, способный к эффективному переносу хиральности, например, с хиральной подложки или с хиральных промоторов на продукт гидрирования было очень много, но результаты редко оправдывали ожидания. Нелишне напомнить, что для того чтобы разработать эффективный катализатор или реагент, нужно хорошо понимать механизм реакции, в которой он принимает участие. У гетерогенных реакций с механизмами до сих пор дела обстоят неважно – головоломно сложная эта штука, поверхность, и как на ней происходят процессы известно только в некотором приближении. Современная наука, конечно, и в этом направлении копает очень интенсивно, но задача чудовищно сложна.
Гомогенное гидрирование в отличие от гетерогенного катализируется комплексами металлов, растворимых в среде реакции, и реакция идет в растворе. Это хорошо, потому что механизмы реакций в растворах изучаются гораздо более точно, и для многих важных процессов уровень понимания происходящего очень велик. Это позволяет все лучше и лучше подбирать структуру катализаторов и достигать выдающихся успехов именно в селективности реакций, в том числе стереоселективности. По этой причине и по многим другим гомогенное гидрирование не общий метод гидрирования (для простых задач все как использовали, так и будут использовать палладий на угле или никель Ренея), а именно специализированный, рассчитанный на специальные задачи, где нужна высочайшая селективность. В асимметрическом гидрировании этому методу нет альтернативы.
Катализатор Уилкинсона
Гомогенное гидрирование открыл не Уилкинсон, и даже гомогенное гидрирование на комплексах родия открыл не Уилкинсон. К моменту выхода главной (или как любят говорить по-английски, seminal work – то есть буквально, работа, ставшая семенем, из которого произросло много нового и хорошего) работы Уилкинсона и сотр. (J. A. Osborn, F.H. Jardine, J. F.Young, G. Wilkinson, J. Chem. Soc. (A), 1966, 1711), уже было описано с полдюжины примеров гидрирования непредельных соединений в растворе в присутствии комплексов кобальта, родия и даже меди. Особая способность самых разных комплексов родия вызывать гидрирование описана прямо накануне Второй мировой войны группой Игути в Японии. Да и сам Уилкинсон чуть раньше уже описал гидрирование на гидридном карбонильном комплексе родия. Сила открытия катализа гидрирования именно комплексом RhCl(PPh3)3, который так и прозвали катализатором Уилкинсона (часто говорят комплексом Уилкинсона, но это не совсем корректно, потому что комплекс уже был известен), состоит в том, что это был первый катализатор гидрирования, который работал с огромной скоростью и показал, что гомогенное гидрирование может быть не хуже гетерогенного в смысле производительности, настолько, что несколько миллиграммов комплекса могут успешно прогидрировать десятки и сотни грамм олефина. В этом случае упреки в дороговизне катализатора и сложностях с возвратом драгоценного родия после реакции становятся не так критичны.
Олефины типа 1-алкенов или циклогексена гидрируются при обычном давлении водорода (глупое слово “обычный” здесь означает, что используется обычная стеклянная посуда, а водород подается в колбу из обычной резиновой камеры или газометра – в этих случаях парциальное давление водорода в сосуде равно атмосферному или превышает его на незначительную величину давления внутри надутой камеры). Реакционную смесь приходится очень хорошо перемешивать – это обычная проблема, когда реакция идет в жидком растворе, а один из реагентов – газ, и скорость растворения газа лимитирует скорость реакции, если реакция быстрая, а это как раз тот случай. В этих случаях водород поглощается с такой скоростью, что кажется, что прибор прохудился и водород хлещет наружу, а раствор разогревается до кипения растворителя (обычно чего-то неполярного типа бензола), – ведь мы помним из курса обычной органической химии, что гидрирование олефинов – сильно экзотермическая реакция. Сам Уилкинсон достигал скорости поглощения водорода до четверти литра в минуту, то есть камера с водородом реально сдувается, как проколотая.
Оценив такую мощь нового катализатора, все почувствовали перспективу за этим направлением и бросились исследовать. Гидрирование на катализаторе Уилкинсона стало и моделью для изучения механизма каталитического процесса.
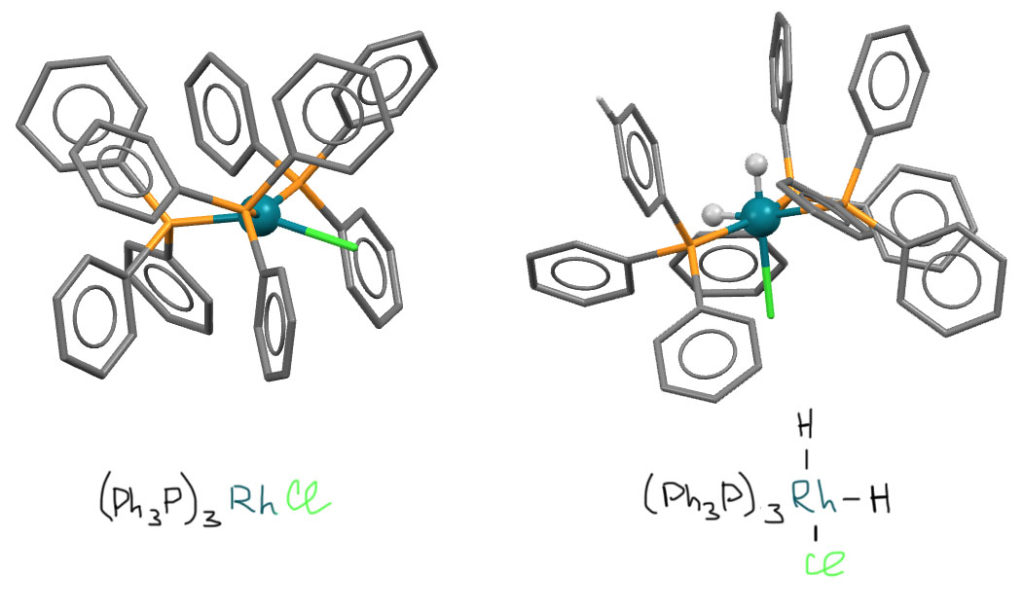
Катализатор Уилкинсона – комплекс Rh(1+), в котором родий имеет конфигурацию d8, то есть это нечто похожее на металлы 10 группы в состоянии +2. Как мы знаем, для этих валентных состояний наиболее характерны 16-электронные плоские квадратные комплексы. Структура катализатора Уилкинсона почти точно соотвествует этому типу, хотя квадрат довольно сильно искажен, почти наверняка из-за стерического отталкивания трифенилфосфинов. Как мы уже не раз видели в других каталитических процессах, это неплохо, потому что приводит к легкой диссоциации одного из фосфинов и образовании 14-e комплекса RhCl(PPh3)2, который-то и является настоящим катализатором. В уже привычных нам терминах исходный комплекс стоило бы назвать не катализатором, а пред-катализатором. Уже сильно координационно ненасыщенный дифосфиновый комплекс легко присоединяет водород с образованием дигидридного комплекса. Это – типичное окислительное присоединение, происходящее за счет back-donation на разрыхляющую σ*-орбиталь молекулы водорода (в современной литературе принято это без стеснения называть диводородом). Вот как выглядят структуры исходного (M. J. Bennett, P. B. Donaldson, Inorg. Chem. 1977, 16, 3, 655-660) и конечного комплексов, но стабилизированного еще одним фосфиновым лигандом. Увы, те комплексы, которые реально работают в каталитическом цикле обычно слишком реакционноспособны, и не могут быть выделены, нуждаясь в дополнении координационной сферы до насыщенности, и то место, которое реальный дигидридный комплекс в каталитическом цикле держит для олефина, пришлось занять. В любом случае, это уже октаэдрический комплекс родия(3+). Для удобства в дигидридном комплексе водороды тоже обозначены шариками.
Итак, сначала водород, потом олефин. Олефин связывает уже комплекс родия(3+), обладающий большим сродством к более донорным олефинам (потому что в связи больше координационного взаимодействия, в котором металл – кислота Льюиса). Посмотрим еще раз на каталитический цикл. Олефин входит, за этим следует миграционное внедрение, и восстановительное элиминирование с оставшимся гидридом. 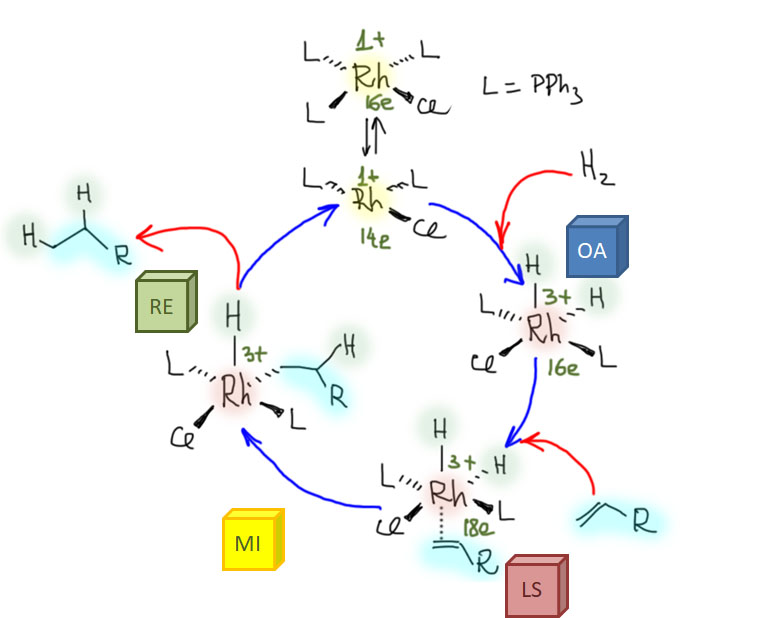
В порядке входа в сферу металла – сначала водород, затем олефин – скрываются главные проблемы этого каталитического цикла.
Во-первых, олефин должен входить в уже очень населенную координационную сферу, в которой уже довольно тесно – поэтому реакция очень чувствительна к размеру олефина. Реакция предпочитает монозамещенные (терминальные) олефины, неплохо справляется с дизамещенными геминальными и цис-олефинами, становится довольно медленной для дизамещенных транс- и очень медленной для тризамещенных олефинов, и отказывается принимать тетразамещенные. Это понятно, в этом нет ничего необычного, и чем больше мы будем узнавать про самые разные реакции,катализируемые комплексами переходных металлов, тем чаще будем обращать внимание на то, что стерический объем субстрата, особенно того, который входит в координационную сферу последним – большая проблема для большинства таких реакций. Кроме этого родий оказывает явное предпочтение олефинам с акцепторным заместителем рядом с двойной связью (олефины типа акцепторов Михаэля, а это говорит о том, что несмотря на степень окисления 3+ во взаимодействии силен вклад back-donation.
Вообще на это стоит обратить особое внимание: легко связываются как олефины с донорными заместителями, так и с акцепторными. Это типичный признак вклада во взаимодействие двух противоположных факторов – и Льюисовой кислотности (угодной донорным олефинам), и back-donation (угодного как раз акцепторным олефинам) – и металл сам решает при взаимодействии, что делать, он умеет и то, и то.
И еще одну вещь любит родий – когда неподалеку от двойной связи есть гетероатом в какой-то группе – спиртовой, эфирной, карбонильной, азот какой-нибудь, фрагмент гетероциклический – такие группы подхватывают родий за одно координационное место и подтаскивают его к двойной связи, буквально тыкая в нее носом – на, типа, гидрируй. Этот эффект называется направленным (directed) гидрированием, и очень распространен в химии переходных металлов.
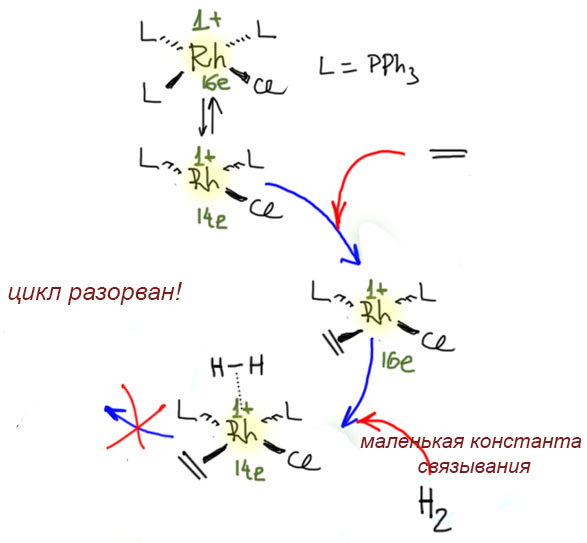
Во-вторых, а что будет, если олефин решит зайти в сферу первым? Будет – облом, потому что такой комплекс уже отказывается окислительно присоединять водород (потому что место занято). И действительно, олефины с большими константами связывания с Rh(1+), успешно конкурируют с водородом. Прежде всего, это сам этилен. Этилен не гидрируется катализатором Уилкинсона. Кроме этого, это всякие диены, и сопряженные и такие несопряженные, которые могут образовать хелат, например, знакомый нам 1,5-циклооктадиен. Все эти молекулы просто блокируют координационную сферу.
Применение гомогенного гидрирования по Уилкинсону
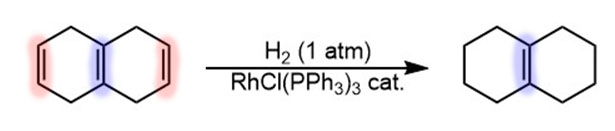
Обычные олефины и ацетилены никто гидрировать так не будет, но в тех случаях, когда важна селективность, гидрирование по Уилкинсону может оказаться очень полезным. Например, так как тетразамещенные олефины не гидрируются, можно легко оставить внутреннюю двойную связь в продуктах восстановления ароматики по Берчу, причем сам Бёрч это и описал (A. J. Birch, K. A. M. Walker, J. Chem. Soc. (C) 1966, 1894–1896), попробовав новый катализатор на куче разных олефинов:
 Такие задачи часто возникают в синтезе сложных молекул, например, стероидов. Эргостерин, например, чисто гидрируется (A. J. Birch, K. A. M. Walker, J. Chem. Soc. (C) 1966, 1894–1896) по одной двойной связи из трех, и не стоит удивляться, почему устояла вроде бы дизамещенная двойная связь в верхней боковой цепи – она транс, а Уилкинсон не любит транс, тем более с рогами по бокам.
Такие задачи часто возникают в синтезе сложных молекул, например, стероидов. Эргостерин, например, чисто гидрируется (A. J. Birch, K. A. M. Walker, J. Chem. Soc. (C) 1966, 1894–1896) по одной двойной связи из трех, и не стоит удивляться, почему устояла вроде бы дизамещенная двойная связь в верхней боковой цепи – она транс, а Уилкинсон не любит транс, тем более с рогами по бокам.
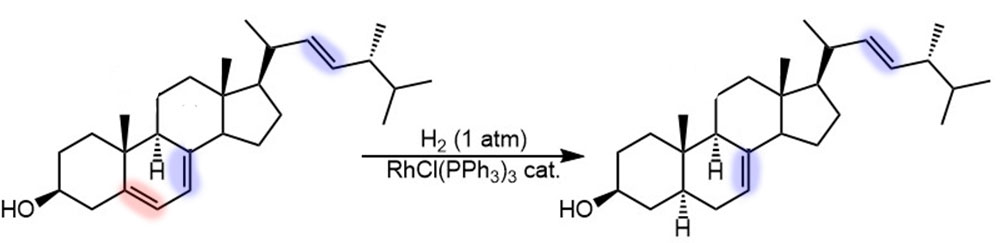
Дополнительно можно и дейтериевые метки вводить, как в этом примере (Li et al, J. Label. Compd. Radiopharm. 2011, 54, 839-846), где катализатор выбирает менее замещенную двойную связь, а дополнительно еще и обеспечивает отличную стереоселективность:
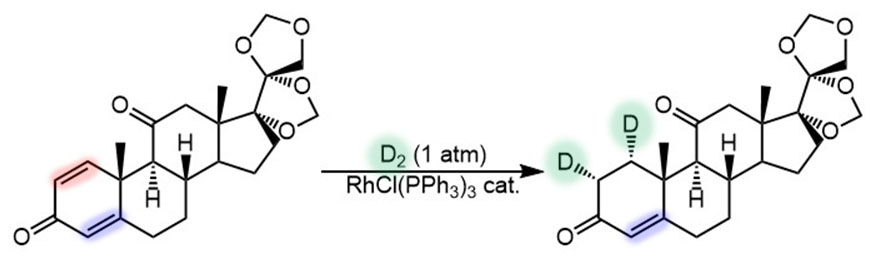
Стереоселективные реакции – общие вещи
В этом разделе я напомню некоторые общие вещи и довольно подробно. Многие всё это знают и могут спокойно пропустить, но я очень хорошо знаю, насколько поверхностно часто к этим понятиям относятся, попадая в результате в плен выдуманных противоречий.
В современной химии нет ничего важнее стереоселективности. Есть несколько вещей столь же важных, но важнее нет ничего. Конечно, далеко не все молекулы имеют стереоизомеры, но те, которые имеют могут похвастаться несравненно большим влиянием в современном мире. Почти всё, что имеет отношение к жизни, в той или иной форме имеет отношение к стереоизомерии. Все химические процессы в клетках организмов обслуживаются чистыми и индивидуальными стереоизомерами, и биохимические процессы всегда производят совершенно конкретные стереоизомеры, сколько бы стереогенных центров ни было бы в их составе – если сто или тысяча, все сто или тысяча будут в конкретной конфигурации. Современный органический синтез не хочет отставать от Природы, и все современные методы синтеза обязательно имеют стереоселективные варианты, если только не относятся к тем немногим типам органических соединений, для которых стереоизомерия не имеет значения.
К сожалению, на 3 курсе мы очень мало внимания уделяем стереохимии, а тому, что называется асимметрическим синтезом, не уделяем внимания вовсе. Химия переходных металлов – современная химия, и в ней стереоселективность играет огромную роль. Пока мы занимались кросс-сочетанием, мы не очень обратили на это внимание, отметив только сохранение конфигурации в реакциях с субстратами винильного типа. Но когда мы переходим к химии олефинов, стереоселективность является к нам, не спросив разрешения, и начинает дубасить в дверь коваными сапогами – всё, от неё больше не спрячешься. И не будем. Пустим-ка её в дверь, она не такая страшная.
Сначала восполним немного пробелы в образовании, оставшиеся с 3 курса. Возьмём реакцию гидрирования, потому что проще ничего не придумаешь, а все закономерности здесь налицо, и если в них сразу разобраться, и в других реакциях будет то же самое.
Итак, гидрируем олефин. Олефин, если в нём нет хиральных заместителей – штука эффективно плоская, нехиральная. Возьмём моно или дизамещённый олефин и прогидрируем. Видим, что и продукт получается нехиральный – в нём нет хиральных атомов углерода, или как чаще говорят сейчас – нет стереогенных центров. Этот термин предпочитают потому, что он не несёт неприятной недосказанности о том, что же такое хиральный атом. Хиральный – значит, не совпадающий со своим отражением в зеркале. Очень понятно, когда это слово применяют к целым молекулам – есть молекула и есть её отражение в зеркале, повертели, и убедились, что они или совпадают или нет. А как отражать и вертеть отдельный атом? Интуитивно понятно как – мы всегда вертим не атом, а молекулу с его участием, настоящую или воображаемую. И если получаем хиральную молекулу, догадываемся, что причиной этого является этот атом, и называем его хиральным. Но есть и более сложные случаи, и тогда у нас начинают появляться сомнения. Вот чтобы этого не было, назовём такой атом стереогенным центром – то есть центром (центр – это то, что можно мыслить как точку; вот атом точно можно мыслить как точку) – центром, рождающим стереохимическую особенность молекулы. Итак, если у нас из нехирального олефина получается насыщенная молекула, не имеющая стереогенных центров, то говорить нам не о чём, кроме обычного – выходов, условий реакции, и т.п.
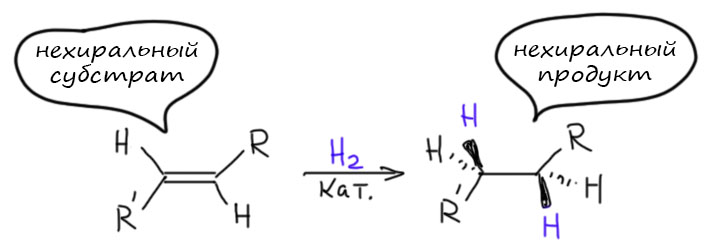
Возьмём теперь олефин, на одном из углеродов которого два разных заместителя (на втором может не быть ни одного, быть один, или два одинаковых). Тогда при гидрировании мы получаем продукт, в котором тот углерод, в котором было в олефине два разных заместителя даст sp3-углерод с четырьмя разными группами – стереогенный центр. Углерод в олефине, превратившийся в стереогенный центр называют прохиральным, то есть предшественником хирального, стереогенного центра. Обратите внимание, что мы пока вообще ничего не говорим про саму реакцию. Она может быть самой обычной – гидрированием на палладии на угле, например, и мы получим оптически неактивный продукт, то есть рацемат – смесь ровно пополам двух энантиомеров. На картинке показан только один из энантиомеров продукта, но это просто для экономии места. Такую реакцию мы назовём нестереоселективной,и мы почти всегда до сих пор только с такими дело и имели. Но возможность получить не рацемат, а один из энантиомеров, в этой реакции уже заложена. Нужно просто подумать, как это сделать.
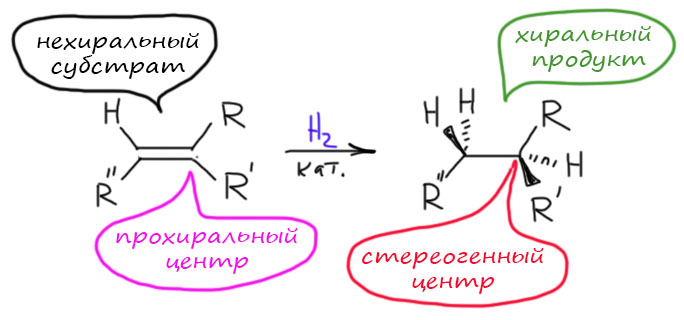
Напишем гидрирование для такого олефина – поскольку второй атом нам ничего не даёт, просто примем его незамещённым для простоты – но показав оба энантиомера. Поскольку R и R’ – произвольные заместители, мы имеем полное право назвать один энантиомер (S), а другой (R). 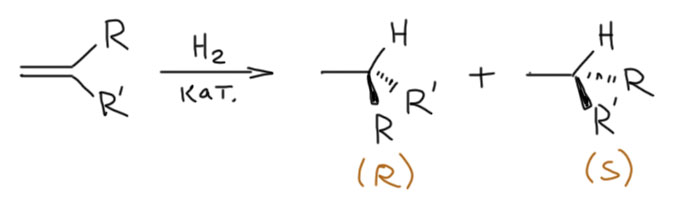
При гидрировании такого олефина хоть с гетерогенными, хоть с гомогенными катализаторам обычного типа (включая Уилкинсона) мы получим рацемат. Что такое рацемат? Это одна молекула одного энантиомера и ещё одна – другого, вот прямо именно энантиомер и его отражение в зеркале, застигнутые вместе? Нет! Как нет?!
Вспомним ещё одно понятие – оптическая активность. Это то же самое, что хиральность, или нет? Эти вещи часто путают, считая синонимами. Но это очень скверная неразборчивость в понятиях. Хиральность – это качество, присущее молекуле. Его можно установить просто по структуре, нарисованной на бумаге в любой проекции, хотя в редких сложных случаях лучше иметь пространственную модель. Хиральность – это просто несовместимость молекулы (или другого материального объекта, но нас это не интересует) и ее отражения в зеркале, причём зеркало можно расположить совершенно любым способом, а значит всегда есть возможность выбрать самый удобный. В принципе, всё это известно, поэтому не будем дальше здесь топтаться на хиральности.
Оптическая активность – свойство не молекулы, а вещества (чистого или в растворе). Оптическая активность устанавливается, в отличие от бумажно-абстрактной хиральности, вполне экспериментальным способом – измерением угла вращения поляризованного света. Если эта величина отлична от нуля, что устанавливается так, как положено при серьёзных измерениях, с помощью матстатистики и соответствующих критериев – вещество оптически активно. Если мы не можем надёжно установить отличие от нуля, то оптически неактивно. В этих измерениях придётся иногда постараться очень сильно, потому что бывает очень слабая оптическая активность, например, такая, которая обусловлена изотопным замещением (дейтерий вместо одного из двух водородов, а ведь можно и один метил с изотопом углерода). Поскольку угол вращения – удельная характеристика, можно взять для измерения раствор с большей концентрацией, но всё равно рано или поздно упрёмся в невозможность измерить с достаточной точностью. И что? И всё.
Так называемые мезо-формы, которые мы иногда, выпендрёжа ради, обзываем внутренними рацематами, всё же никакие не рацематы, а просто нехиральные молекулы, содержащие стереогенные центры противоположной конфигурации. Поэтому молекула не может быть рацемической. Рацемическим может быть только вещество (а смесь – это вещество? конечно – совершенно нигде не сказано, что вещество обязательно состоит из молекул одного сорта, более того, это принципиально недостижимо, а следовательно и неверно). И когда в задачах в курсе органической химии спрашивают про какую-то структуру, является ли она оптически активной, имеют в виду, не структуру и не молекулу (про них корректнее было бы спросить, являются ли они хиральными), а вещество, соответствующее этому соединению. Химия, в этом и всех подобных случаях – злобная гадина, испытывающая нас на профпригодность самым циничным способом, задавая бессмысленные вопросы и ожидая от нас осмысленного ответа там, где правильнее было бы послать куда подальше. Она всегда делает вид, что спрашивает про молекулы, но имеет в виду всегда вещество, и всегда совершенно конкретное. Если чистое, то конкретно какое – жидкость, газ, твёрдое, это важно и этим нельзя пренебречь. Если раствор, то определённой концентрации или диапазона концентраций, и выходить далеко нельзя. Мы всегда, если хотим оставаться химиками, то есть представителями одной из важнейших естественных наук, должны понимать, что всё устанавливается в воспроизводимом эксперимента, а не в голове. Поэтому вопросы типа такого, – ну, для молекулы нельзя установить оптическую активность, а для ста или тысячи молекул – можно? Ведь это уже вещество, наверное. Наверное. Но есть ещё такая штука, как точность эксперимента, и это не абстракция, а вполне конкретный, почти железобетонный предел для бесплодных фантазий, особенно если учитывать то, что точность эксперимента определяется не криворукостью экспериментатора, как некоторые думают, а вполне физическими принципами работы измерительной аппаратуры и статистической природой объекта измерения и самого процесса измерения. И химику никогда нельзя забывать про порядок величины числа Авогадро.
Возвращаемся к реакции гидрирования. На реакции мы ещё раз и очень наглядно увидим разницу между хиральностью и оптической активностью. Представьте себе, что гидрируется ровно одна молекула. Что получится? Один энантиомер. Либо один, либо другой, но один. Если бы мы смогли повторять реакцию по одной молекуле, в каждом “эксперименте” у нас получался бы один энантиомер, но случайно, либо один, либо другой. Мы бы поняли, что образование конкретного энантиомера это типичное случайное событие, и это равновероятно, как монетку кидать. А теперь сделаем “эксперимент” сразу с десятью молекулами. Получится ли у нас “рацемическая смесь” – ровно 5 одного и 5 другого. Возможно, но если вы ещё помните теорию вероятностей, то поймёте, что вероятность такого результата хотя и больше чем любого из других результатов, но уже ненамного больше, чем близких результатов (4 одного вида и шесть другого – а ведь это уже не “рацемат” никакой!). А если возьмём сто молекул, то событие 50-50 станет уже маловероятным на фоне большого количества близких вариантов с небольшой разницей. Из тысячи молекул вариант 500-500 станет уже просто маловероятным. И так далее. Эта дурацкая игра показывает, что рацемат (рацемическая смесь) ни в коем случае не предполагать штучное равенство числа энантиомеров, – это просто такая смесь, для которой мы не можем достоверно определить преобладание одного энантиомера над другим, а значит и имеем оптическую активность (вращение) равной нулю в пределах точности измерения. И если бы нам предоставилась возможность поштучно посчитать количество энантиомеров каждого вида в каком то реальном образце реального рацемата, мы, почти наверняка, обнаружили бы вполне ощутимое неравенство. Такие рассуждения можно было бы считать глупой игрой с теорией вероятностей, если бы не одно обстоятельство – теория вероятностей предполагает, что число испытаний всегда достаточно велико, чтобы всё усреднилось к теоретическим цифрам. Но реальный мир может не дать такой возможности, и остановиться на одном случайном событии, дальше произведя всё остальное из этого единичного испытания. Мы ведь до сих пор не знаем, откуда на Земле взялась жизнь со всей своей оптической активностью, но большинство исследователей, размышляющих на эту тему, сходятся к мысли, что источником жизни явилось какое-то случайное единичное, а следовательно и чрезвычайно маловероятное, событие, или даже цепь таких событий.
Но у нас пока эксперимент не так смел и уникален – мы просто пытаемся понять, откуда может взяться оптическая активность в реакции, например, гидрирования. И если хиральность может взяться из двух нехиральных молекул, то оптическая активность, если мы поняли, что это такое, не может. Можно даже сформулировать такое правило:
Если все реагенты и вспомогательные вещества, то есть всё то, что мы пишем или подразумеваем слева от стрелки – растворители, катализаторы, добавки любой природы и назначения, – над и под стрелкой реакции оптически неактивны, то и результат такой реакции всегда будет оптически неактивен.
Обратное правило тоже справедливо, но с одним важным но – оно работает только как необходимое условие. Но не гарантирует результата.
Для того чтобы продукт реакции имел оптическую активность необходимо чтобы хоть что-нибудь из того, что мы рисуем слева, сверху и снизу от стрелки реакции – исходные вещества, реагенты, катализаторы, растворители, добавки – было оптически активным.
Оптическая активность не возникает из ничего, а как будто передаётся (как коронавирус, чёрт бы его побрал, или другая какая зараза) от вещества к веществу.
Для обозначения этого явления используют разные термины, чаще всего два – асимметрическая индукция (asymmetric induction) и перенос хиральности (chirality transfer). В общем, это одно и тоже, и дело вкуса, какое из них употреблять – вполне можно и оба, рассматривая их как синонимы. Слово “индукция” не очень нравится людям, строго следящим за смыслами, потому что порождает ненужную ассоциацию с тем, как это происходит в физике, например, с электромагнитной индукцией. В физике это происходит через пространство, как будто через ничего (это ничего “наполнено” электромагнитным полем, но кто ж его видел). Но асимметрическая индукция никогда не происходит через пространство – для него нужна совершенно конкретная, почти осязаемая сближенность молекул – индукция происходит через стерические взаимодействия, делающие альтернативные, про-(S) и про-(R) направления реакции, порождающие индивидуальные энантиомеры, неравными по энергии активации, а значит и по скорости. И чем теснее такие взаимодействия, тем больше разница, тем выше энантиоселективность. Впрочем, перенос хиральности тоже не безупречен, особенно если вдуматься – кто куда чего перенёс, чёрт знает – это, если вдуматься, намного более литературная метафора, чем строгий термин, тем более что мы уже разобрались, что хиральность это свойство молекул, а не веществ, и она как раз очень даже может возникнуть из ничего. Но где вы видели строгие термины в химии? Живём пока с этими, а вы или ваши потомки наверняка придумаете что-нибудь получше.
Раз мы уже разобрались с тем, что оптическая активность – свойство вещества, нам будет легко понять, что это переменная величина – она изменяется от отсутствия оптической активности до некоторого максимума для каждого вещества, представленного парой энантиомеров. Опять таки в отличие от хиральности, у которой только два дискретных состяния – есть или нет. Понятно, что отсутствие соответствует рацемической смеси, а максимум – чистому энантиомеру. Для характеристики оптической активности используют величину оптической чистоты или энантиомерного избытка (enantiomeric excess, сокращённо ee). Это очень простая и очевидная величина, но для неё нужно знать удельное вращение любого чистого энантиомера интересующего вещества, и при измерении удельного вращения образца суметь убедиться в том, что с химической точки зрения образец чист от примесей, которые могут оказаться оптически активными и исказить результат измерения.
Дальше всё очень просто и интуитивно. Если у вас в образце пополам энантиомеров, это рацемическая смесь, оптически неактивная. Если чистый энантиомер, то оптическая чистота равна 100%, а угол вращения (по модулю) максимален. А если, например, у меня 80% одного и двадцать другого? Оптическая чистота 80%? Нет, ведь там 20% другого энантиомера, а значит 20% этого образуют с тем рацемическую смесь, оптически неактивную, и только 80-20=60% будут вращать плоскость поляризации на 60% от максимального значения (по модулю). Оптическая чистота 60%. И вот почему это называют энантиомерным избытком – избытком над рацемической смесью. 80% большего энантиомера образуют 60%-ный избыток над рацемической смесью. И нас интересует именно это, потому что только избыток энантиомера над рацемической смесью порождает оптическую активность. Нет избытка – нет оптической активности.
И это очень легко измерить и оценить. Экспериментально, как видим, это просто доля от максимума (по модулю), выраженная в процентах. Не забываем, что вращение – удельная величина, измеряемая для раствора известной концентрации, и приведённая к единичной концентрации.
- Померили вращение,
- определили знак,
- нашли чистый энантиомер, имеющий такой же знак
- померили его вращение или посмотрели в справочнике (справочники иногда врут, поэтому померить лучше)
- поделили вращение образца на вращение чистого энантиомера, умножили на сто
- убедились, что полученная величина положительна, больше нуля и меньше ста
- вуаля – энантиомерный избыток, можно радоваться, если велик, или сокрушаться, если мал
- повторили измерения столько раз, сколько нужно, чтобы величина была статистически достоверна
Энантиоселективность
Самый простой и распространённый случай – исходный олефин оптически неактивен, и для простоты примем, нехирален. А разве это не одно и тоже? Нет, мы же уже всё разобрали – оптическая активность – свойство вещества. Представьте себе олефин, в составе которого есть стереогенный центр. Такой олефин хирален. Возьмём этот олефин в виде рацемата. Тогда такой олефин оптически неактивен. Это важный случай, но мы его разберём во вторую очередь. Сначала представим себе, что олефин нехирален. Мы уже сто раз писали пример такой реакции.
Поскольку в самом олефине хиральности нет, ее придётся переносить извне, с компонентов реакционной смеси. В принципе, любой компонент реакционной смеси может переносить хиральность, но эффективность этого процесса обычно очень мала. В прошлом веке исследователи топтались на этой проблеме несколько десятилетий, пробуя всё подряд. Продукт получался оптически активным, но с очень небольшой оптической чистотой (энантиомерные избытки порядка нескольких процентов, иногда даже десятка-полутора процентов. Поначалу это было интересно, но быстро стало понятно, что для синтеза это бесполезно. Понемногу сложилось представление о том, что эффективный перенос хиральности можно ждать только если компонент реакционной смеси очень сильно взаимодействует с исходным на тех стадиях, которые непосредственно ведут к продукту. Если оба исходных, олефин и водород, нехиральны, то такому условию соответствует только катализатор, очень сильно взаимодействующий с исходными.
Особенно важно взаимодействие с тем реагентом, который содержит прохиральный центр – этот реагент должен суметь образовать с катализатором хиральный интермедиат с максимально определённой структурой – желательно, чтобы там ничего не вертелось, и чтобы те фрагменты катализатора, которые определяют его хиральность, и прохиральный атом олефина, были максимально сближены и жёстко закреплены относительно друг друга. Как это реально работает, мы увидим дальше на примере катализатора Ноулза – хиральность катализатора там определяется не монодентатным, а бидентатным лигандом, образующим хиральный 5-членный хелатный цикл – такой цикл прочен, и это ровно то, что надо. Более того, и олефин годится не любой, ведь в дигапто-комплексе олефин крутится вокруг оси связи, а должен сидеть как вкопанный. И этого можно добиться, если в олефине есть ещё какая-то группа, способная стать лигандом, а всязывание олефина с такой группой – образованием хелата. Из этого следует довольно грустный вывод – реакции энантиоселективного гидрирования редко бывают хоть немного общими и пригодными для разных олефинов, гораздо чаще их (в данном случае это означает подбор эффективного катализатора) подбирают под конкретные важные олефины, довольно специфические по структуре.
А вот до открытия Ноулза много экспериментировали с монодентатным лигандами, содержащими так называемые хиральные подвески (chiral pendants), то есть просто какие-то доступные хиральные фрагменты в виде заместителей. Ну, например, брали природный хиральный спирт ментол, и использовали его, чтобы получить хиральный заместитель ментил, например, просто на фосфине. И такой катализатор работал плохо, потому что а) олефин в дигапто-комплексе крутится относительно металла, и заставить его не крутиться можно только создав жёсткую, конфигурационно стабильную, и очень тесную координационную сферу, а монодентатный лиганд делать этого не может; б) собственно хиральный центр далеко от реакционного центра (атома металла), и такой центр слабо влияет на разницу направлений реакции про-(S) и про-(R) – скорости реакций, ведущих к индивидуальным энантиомерам различаются мало, и что вы тогда хотите?
Энантиоселективность конкретной реакции характеризуют оптической чистотой продукта, величиной энантиомерного избытка, ee. То есть, для каталитической реакции указывают материальный выход от теории и энантиомерный избыток. Будьте внимательны, когда смотрите статьи с такими данными и схемы реакций в них. Величина в процентах, указанная под стрелкой или продуктом может быть и тем, и тем.
Диастереоселективность
Диастереоселективность – преимущественное образование одного из диастереомеров в реакции. Это гораздо более широкое понятие, чем энантиоселективность, и даже не всегда связанное с оптической активностью. Разновидностей диастереомеров довольно много (посмотрите на другом сайте, если забыли). Например, это цис- и транс-изомеры олефинов или замещённых алициклов. И, например, если мы найдём катализатор гидрирования дизамещённых алкинов до алкенов, могут получаться как цис- так и транс-олефины, и мы будем искать стереоселективный метод получения тех или других, но будем брезговать методом, дающим смесь цис и транс. Это – типичный пример диастереоселективности.
Интересно, что диастереоселективность по аналогии с энантиоселективностью принято характеризовать величиной диастереомерного избытка, de. Просто по аналогии, никакого смысла. Ну вот представьте себе, что у вас есть смесь 80 морковок и 20 прищепок. И вы говорите, что получили избыток в 60 морковок. Избыток над чем? Над загадочной смесью из 20 морковок и 20 прищепок. Я бы ещё немного понял, если бы была смесь 80 кочерыжек и 20 кроликов. Тогда говорить об избытке в 60 кочерыжек хотя бы немного имеет смысл, потому что 20 кроликов схрумкали бы 20 кочерыжек, и правда остался бы избыток в 60 кочерыжек, по крайней мере пока кролики вновь не проголодались бы. Вот так же и с диастереомерным избытком. Получили вы, скажем, смесь 80% цис-алкена и 20% транс-алкена. Говорите, что у меня (у вас, а не у меня), типа, диастереомерный избыток в 60% цис. Избыток над чем? Над смесью 20% цис и 20% транс. А что это за смесь, чем она интересна? Да ничем, просто смесь бесполезная. Ведь диастереомерные цис- и транс-алкен – совсем разные соединения, с разными свойствами, почти ничем друг с другом не связанные. Смешать их – то же самое, что смешать другие разные соединения. Эта смесь не связана ни с каким интересным свойством, и нет никакого свойства, с помощью которого вы сможете быстро это определить. Вы просто измерите с помощью хроматографии или ЯМР содержание каждого из диастереомеров. И справедливости ради скажем, что большинство исследователей так и напишет – получили смесь цис/транс = 80:20, не заморачиваясь никакими избытками. Но манера вместо этого писать диастереомерный избыток весьма распространена особенно среди исследователей, работающих со стереоселективностью различных типов, и им кажется, что диастереомерный избыток и энантиомерный избыток образуют такую красивую симметричную пару, не замечая того, что одна характеристика отлично обоснована и очень полезна, а вторая – просто такие понты, весьма сомнительные. Но раз мы будем это встречать, должны знать, что это такое.
Подойдём ближе к стереоселективному гидрированию. Давайте гидрировать олефин, уже содержащий хиральный атом в стороне от двойной связи, причём энатиомерно чистый. Вот что мы получим в общем виде: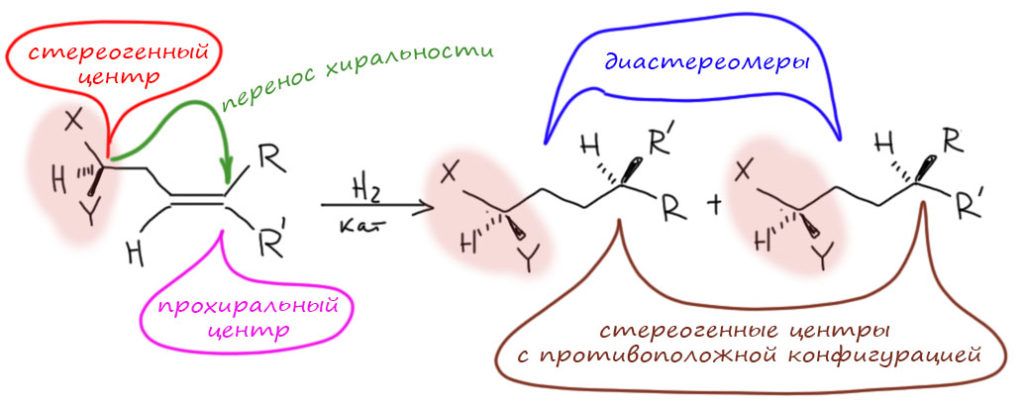
Если у нас больше нет в реакции ничего оптически активного, мы всё равно получим при гидрировании не просто оптически активный продукт (оптическая активность гарантирована наличием не участвующего в реакции непосредственно хирального центра, если только не случилась какая-нибудь случайная рацемизация, что бывает и нередко), а продукт, состоящий из двух диастереомеров, причём почти наверняка в неравных количествах. Одного будет больше чем другого, мы априори не знаем какого и насколько.
Откуда взялось неравенство на новых стереогенных центрах? Произошёл перенос хиральности с уже имеющегося, внутрмолекулярный. Эффективно ли это? Бывает что да, но обычно нет, особенно в нециклических молекулах, где есть вращение, много конформаций, и ничего не закреплено. И чем дальше этот старый центр, тем, как правило, хуже, понятно почему.
Просто для примера примем, что старый центр имеет конфигурацию (R). Новые – (R) и (S). Тогда в продукте мы получим два диастереомера (R,R) и (R,S). Допустим, опять примера ради, что первого больше чем второго, пусть будет (R,R) – только не нужно вкладывать в это никакого смысла, конфигурации центров никак прямо не связаны, и чтобы в конкретном случае понять, как происходит перенос хиральности, нужно строить пространственные модели переходных состояний и пытаться понять, почему стали разными вероятности атаки с разных сторон. Пусть первого будет 60%, а второго 40%.
Зададим вопрос, что такое разность между выходами диастереомеров – 60-40=20%, и что за селективность мы наблюдаем – энантио или диастерео? Довольно очевидно, что вторую, и эта разность – это именно диастереомерный избыток, de = 20%.
А теперь зададим вопрос, какова оптическая чистота продукта. Вот это уже вопрос не такой простой, потому что у диастереомеров удельное вращение различно, и никаких простых соотношений нет – они вполне могут, например, иметь один знак. Это больше не энантиомеры, больше нет рацемической смеси, и нет очевидного смысла слова избыток, хотя, безусловно, если мы знаем вращение чистых диастереомеров этого типа, можем сделать линейную зависимость и опрелелить соотношение интерполяцией. Но если уж мы нашли это соотношение и определили разность, мы имеем полное право использовать ее как меру оптической чистоты, и даже как энантиомерный избыток. Энантиомеров здесь нет, но энантиомерный избыток мы имеем право использовать, понимая его как разность между выходами продуктов с противоположной конфигурацией нового стереогенного центра. В этом конкретном случае он будет равен диастереомерному, хотя бывают случаи сложнее, когда это не так.
И ещё один эксперимент. Проведём гидрирование того же хирального субстрата, но с хиральным катализатором. Получим опять два диастереомера. Откуда идёт перенос хиральности – изнутри или из катализатора? В общем случае чёрта с два мы это поймём, потому что эти два источника асимметрии не обязаны позитивно взаимодействовать, и вполне могут даже мешать друг другу. Катализатор вполне может даже играть в пользу другого диастереомера. Но мы, естественно, постараемся найти такой катализатор, чтобы получить оптическую чистоту получше. И если сможем, то это значит, что мы успешно переключили перенос хиральности с одного источника на другой.
На этом месте пока поставим точку.
Асимметрическое гидрирование
Вот как выглядит предкатализатор в гидрированию по Ноулзу – комплекс родия с хиральным дифосфином DIPAMP и вспомогательным лигандом COD. Поскольку все лиганды здесь нейтральные, то комплекс заряжен и имеет противоион: [(DIPAMP)Rh(COD)]BF4. Показан только комплекс родия без противоиона, зато в трех проекциях:
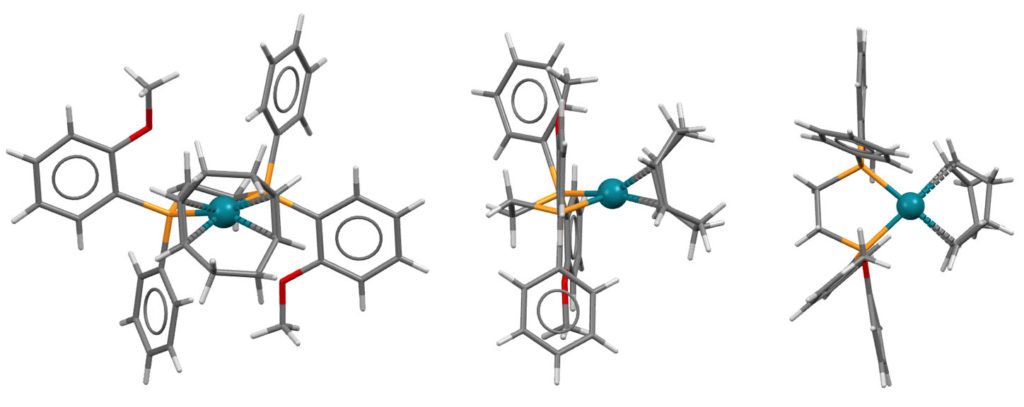
Активация предкатализатора происходит очень просто – эта штука сама себя гидрирует и циклооктан отваливается. Остается приблизительно вот что:
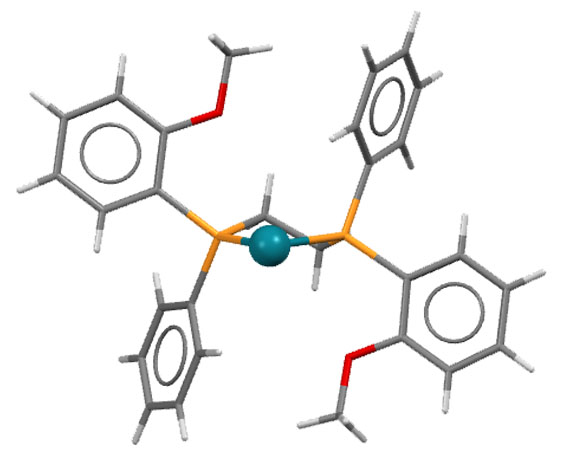
Очень хорошо видно, что хиральный лиганд создает несимметричное окружение вокруг атома родия: лево не такое как право, а верх не такой как низ. Очень важно то, что источник асимметрии находится в непосредственной близости от атома родия, на котором будет происходить собтвенно реакция гидрирования в виде последовательности стадий, во время которых реагирующие лиганды расположатся так, чтобы минимизировать стерические взаимодействия с имеющимся лигандом.
И вот когда мы смотрим на каталитический цикл гидрирования, мы это и видим.
1. К комплексу родия с хиральным фосфином сначала пристыковывается субстрат – олефин, но имеющий рядом с двойной связью функциональную группу, которой жестко закрепляется на атоме родия как хелатный лиганд.
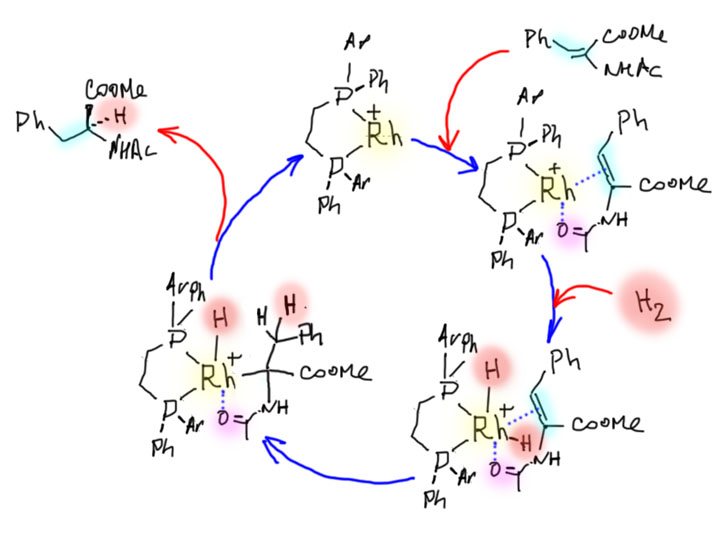
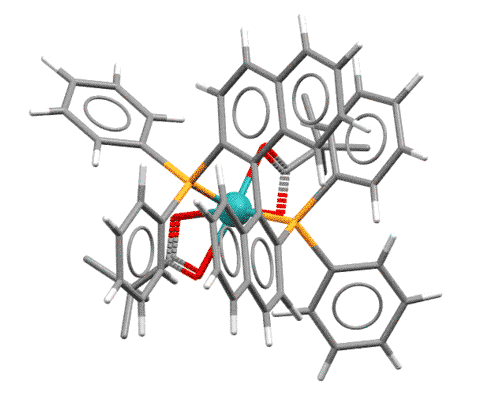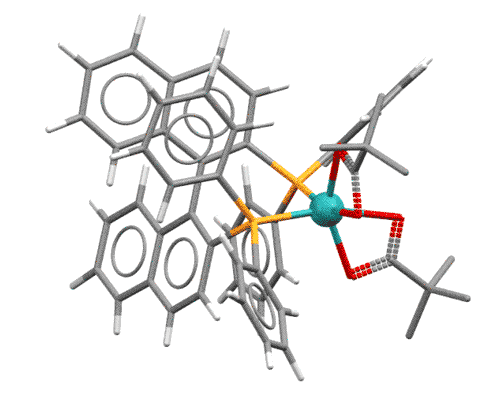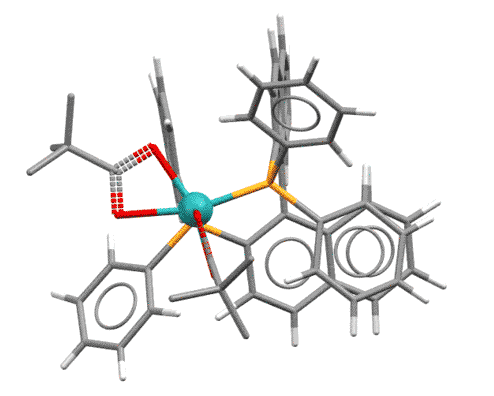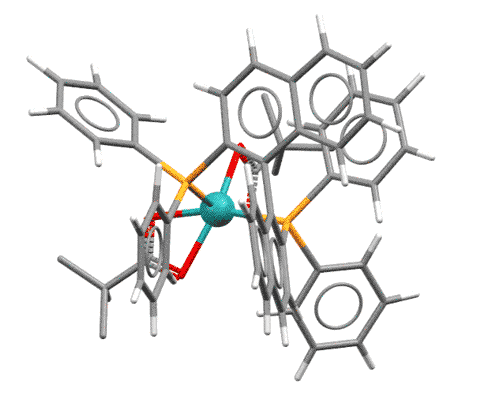
В 2005 году такой комплекс был выделен, и его структура определена (D.Heller et al, Angew. Chem. Int. Ed. 2005, 44, 1184 –1188). Строго говоря, это исследование сделано не на самом важном субстрате асимметрического гидрирования, альфа-ацетиламиноакрилате, дающем выход к знаменитой леводопе, а на очень похожей молекуле, бета-ацетиламиноакрилате. Так часто бывает в сложных исследованиях – приходится заменять один субстрат другим, потому что с тем первым никак не удается что-то там закристаллизовать или выделить. А с этим удается. И на нем видно самое важное, что определяет успех всего процесса. И это позволяет сделать обобщение. Если в последующих исследованиях получится уточнить или даже переписать механизм, то и флаг в руки будущим исследователям, так и только так устроена серьёзная наука.
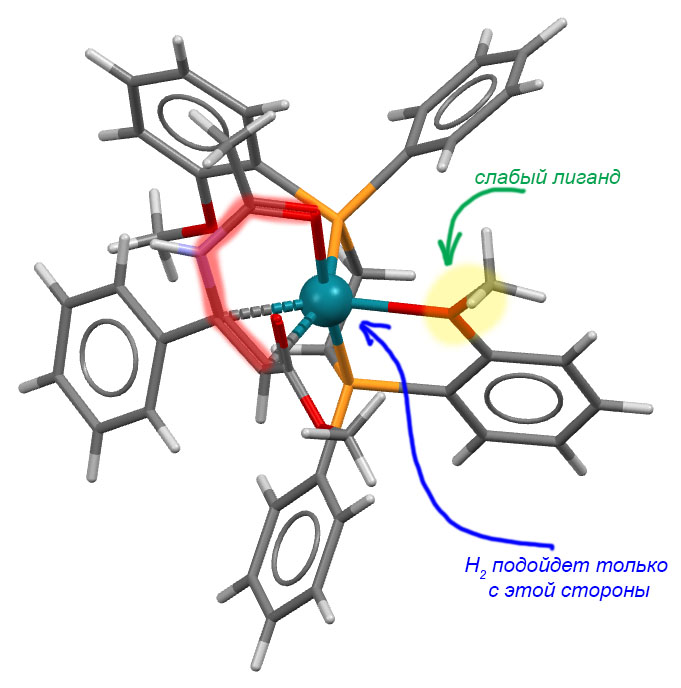
Структура, конечно, очень громоздкая, но попробуем в ней все же разобраться. Обращаем внимание на несколько вещей. Для удобства я подсветил субстрат (не весь, а только то, что образует хелатный цикл) красненьким. Видим, что он закрепился вполне определенным способом. Почему? Во-первых, потому что своими непростыми формами вписался в свободное пространство, оставленное лигандом в координационной сфере. Во-вторых, потому что такова энергетика комплекса – кислородом он зацепился в транс-положении к атому фосфора, и скорее всего, этой конфигурации соответствует меньшая энергия. Чтобы это узнать поточнее, нужно с помощью квантово-химических расчетов очень высокого качества рассчитать все возможные конфигурации комплекса. Кто-то, вероятно, это даже сделал, но нам это не нужно – нам нужно понять принцип, а принцип в том, что субстрат жестко и строго определенным образом закрепляется на атоме родия с симметрическим лигандом. А почему мы в этом уверены – может в реакционной смеси и другие комплексы образуются? Потому что мы знаем, что в данной конкретной реакции с данным конкретным лигандом и данным конкретным субстратом получилась очень высокая энантиоселективность – значит геометрия всех комплексов по дороге цикла была строго определена.
А почему тогда не 100%? А вот это непростой и весьма принципиальный вопрос. Причин может быть несколько. Первая – возможно, комплекс с другой конфигурацией тоже образуется, но имеет более высокую энергию, что по всем законам термодинамики (связывание субстрата на металле это равновесие) приводит к равновесию между двумя комплексами, “правильным” и “неправильным” с константой равновесия на стороне “правильного”, но “неправильный” комплекс забирает часть циклов и приводит к неправильному энантиомеру. Вторая – в реакции участвует комплекс родия или вообще без хирального лиганда, или с ним, но в монодентатном связывании с металлом, так сказать, с оторвавшейся ногой – например, потому что один из фосфинов окислился примесью кислорода в фосфиноксид. Так бывает и нередко – погрешности в эксперименте бывают у всех. Но нужно понимать, что в данном случае мы имеем дело не с результатом одного опыта, а с выводами огромной работы по оптимизации всего на свете для внедрения системы в промышленную эксплуатацию. И это, почти наверняка, среднее из сотен или тысяч оптимизированных экспериментов. Поэтому, дело, скорее всего, в первой причине – в неабсолютной дискриминации “неправильных” интермедиатов. В таких случаях обычно делают простую вещь – проводят реакцию при более низкой температуре, и если дело в конкуренции более стабильного (“правильного”) и менее стабильного (“неправильного”) комплексов, то при пониженной температуре более стабильного комплекса станет относительно еще больше, и селективность должна улучшиться. Так обычно и бывает, но не надо забывать, что при понижении температуры падает и скорость реакции, и лишних 2-3% энатиоселективности придется слишком долго ждать, а если речь заходит о промышленном процессе, то время, как известно, деньги, издержки, убытки. Находят компромисс между скоростью и энантиоселективностью. Тем более, что если энантиоселективность уже велика, то дополнительную очистку основного энантиомера часто можно достичь банальной перекристаллизацией, потому что у чистого энантиомера и рацемата разные кристаллические упаковки, а следовательно, может быть и разная растворимость.
Еще раз возвращаемся к комплексу. Итак, субстрат закрепился. И мы видим еще одну забавную штуку – один из метоксифенильных заместителей фосфина подцепил родий своим кислородом. Это дает дополнительную фиксацию несимметричного окружения в координационной сфере и почти безусловно добавляет жесткости в фиксации субстрата. Но то же самое дает и еще один интересный эффект: нам же теперь нужно засунуть молекулу диводорода. А куда ее засунуть – с этой метокси-группой комплекс вообще кажется координационно-насыщенным. Это так, но это тот самый слабый, временный, неявный лиганд, который просто “держит место”, как это часто бывает и в жизни. Когда на горизонте обозначается водород с гораздо более серьезной константой связывания (а откуда мы это знаем? – из общих соображений о свойствах разных металлов, а также просто потому что реакция идет), то он заменяет метокси-группу и поэтому тоже входит в сферу с совершенно конкретной стороны, и когда происходит окислительное присоединение, то два гидрида тоже образуются с этой стороны и в цис-расположении.
Возвращаемся к каталитическому циклу. Чтобы далеко не ходить, повторим его здесь еще раз, и зарисуем все же для альфа-ацетиоаминоакрилата, посчитав, что основные особенности закрепления субстрата на металле работают похожим образом.
Теперь мы видим, как точно все закрепилось в координационной сфере. Происходит миграционное внедрение, первый гидрид мигрирует на дальний атом углерода, и пока никакой новой хиральности не образовалось. Но это иллюзия – ведь одновременно родий сел σ-связью на ближний углерод – и сделал это совершенно определенным способом – с одной стороны, то есть именно сейчас произошел перенос хиральности – атом углерода, соединенный с родием, имеет точную стереохимическую конфигурацию. И когда происходит последний акт – восстановительное элиминирование, оно происходит с той же стороны, с сохранением конфигурации. В этот момент фиксируется стереохимический результат.
BINAP – хиральный лиганд: революция в энантиоселективном гидрировании
Пока просто посмотрим, как выглядит пред-катализатор для гидрирования по Нойори. Очень хорошо видна та же черта, что и у комплекса, использованного в гидрировании по Ноулзу – и там, и там, лиганд дифосфинового типа, образующий прочный хелат с металлом, в котором оставшаяся часть координационной сферы металла приобретает сильную асимметрию.