
Вопросы и ответы
Я получил много вопросов по лекциям. Это очень хорошо и я очень благодарен всем, кто их присылает. Это очень важно, чтобы содержание лекций становилось более содержательным, понятным и, по возможности, лишённым глупых ошибок. Вопросов так много, что я немного озадачился, что с ними делать. Некоторые вопросы очень конкретны, и я просто вношу уточнения в тексты слайдов и пояснений. Но другие вопросы затрагивают гораздо более общие проблемы и уточнением не отделаешься. Рано или поздно я по следам таких вопросов сделаю значительный update соответствующих тем, но это потребует много времени. Поэтому я решил завести такую страничку для быстрых и коротких ответов, и эта страничка будет потом мне напоминать, что нужно ещё добавить на сайт.
Пожалуйста обратите внимание, что я копирую сюда вопросы и ваших писем анонимно. Если кто-то из вас хочет, чтобы вопрос задавался явно от вашего имени, вы должны дать мне разрешение упоминать вас как автора вопроса. Все вопросы великолепны и содержательны, и быть авторами таких вопросов точно не зазорно.
Вопросы 2023 года
Пока это скорее напоминалки для меня самого. Я немного позже решу, ответить ли здесь, или просто включить некоторый апдейт в лекции.
Бесфосфиновый катализ
Ответ: ждите, разбираются…
Гетерометатезис
Ответ: ждите, разбираются…
Внутримолекулярный Хек
Ответ: ждите, разбираются…
Анти-элиминирование в каскадах с карбопалладированием.
Еще я нашел серия интересных работ с формальным анти-карбопалладированием (одна из первых работ по этой теме — pawliczek2015), в которых авторы предполагают, что при определенных условиях возможна изомеризация винил-палладиевого интермедиата.
Ответ: ждите, разбираются…
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Вопросы 2020 года
Окислительное присоединение
И в принципе, какие связи могут претерпевать ОП? Как это определяется? Анализом расположения той самой разрыхляющей орбитали нужной нам связи? Почему, например, не происходит окислительное присоединение металла по связи С-Р в фосфиновом лиганде? Или, например, если мы берем какой-нибудь арил галогенид, то почему ОП происходит исключительно по связи С-Галоген, а не происходит, например, по 5 оставшимся связям С-Н? Ну и так далее. Хочется научиться понимать по каким связям может происходить ОП, а по каким – нет. Вот прям смотреть на структуру и видеть это.
Ответ: ОП – это окислительное присоединение, а ВЭ – восстановительное элиминирование. Сразу прокомментирую последний пассаж: “Хочется научиться понимать … прям смотреть на структуру и видеть это”. Если бы в химии всё было так просто, скорее всего в ваших услугах в последующие десятилетия 21 века химия уже не нуждалась бы, и приём на химический факультет пришлось бы закрыть. Химия пока ещё не превратилась в точную науку и никогда не превратится. Химия – это наука естественная, что означает, что она экспериментально пытается немного разобраться в устройстве Природы. И как и раньше, так и в последующие десятилетия, а скорее всего, и столетия, химия будет в значительной степени искусством делать разумные выводы из неполных данных и очень приблизительных “теорий”. Это хорошая новость, потому что для такой науки потребность в профессиональных учёных и практиках будет только расти.
Тем не менее, с окислительным присоединением разобраться можно. Окислительное присоединение определяется тремя факторами, два из которых присутсвуют всегда, а третий – только иногда.
- окислительному присоединению легче всего подвергается связь, которую легче разорвать восстановлением за счёт переноса электрона, то есть или электрохимически (на катоде), или действием самых электроположительных металлов, а лучше всего прямо электронов (раствор щелочных металлов в жидком аммиаке).
- для связей углерода с другими элементами (а окислительному присоединению отлично подвергаются и связи между двумя не-углеродами, но это уж совсем отдельная история) очень важна та часть, которая висит на втором элементе, не углероде. Эта часть или должна быть выдающейся уходящей группой типа трифлата (просто хорошие, отличные, превосходные и пр. уходящие группы не катят – нужна именно совершенно выдающаяся). Или она должна быть очень хорошим лигандом для того конкретного металла, который взялся эту связь разорвать. Это обусловлено тем, что металл таким образом – связывая очень хороший лиганд – получает достойную компенсацию за свои усилия. Для разных металлов хорошие лиганды различны. Палладий предпочтёт тяжёлые галогены и брезгует всем кислородным или азотным, а никель может очень порадоваться более жёстким лигандам. Рутений же так любит водород, что может в некоторых обстоятельствах наехать и на связь C-H.
- третий фактор может сработать тогда, когда два первых говорят “нет”. Это то, что мы обсуждали в последней лекции – направленная реакция, когда координирующий заместитель направляет металл на ближнюю связь, даже если в обычных условиях металл и внимания бы на неё не обратил.
Как узнать самые слабые связи в молекуле, те, которые с большей вероятностью будут разорваны одноэлектронным восстановлением. Нужно посмотерть на электронную структуру, сделать расчет каким-нибудь приличным квантово-химическим методом или использовать аналогию. В расчете мы увидим молекулярные орбитали. Нас будут интересовать занятые орбитали. Нужно ли обязательно брать ВЗМО? Нет. Особая роль именно ВЗМО в реакциях проявляется только тогда, когда эта орбиталь в пакете орбиталей обособлена и так одиноко торчит сверху. Так бывает обычно в пи-системах, кратных связях, ароматических системах и т.п. Обратите внимание, что нам это принципиально не нужно, так как мы нацеливаемся на простые связи, σ-связи – только такие связи подвергаются окислительному присоединению. Вместо этого нас должны заинтересовать несколько занятых орбиталей, находящихся на диаграмме близко к ВЗМО. В реальных данных расчётов вы без труда это увидите – на самом верху занятых орбиталей обычно находятся несколько (две, три, четыре) близколежащих орбиталей. Чем сложнее молекула, тем больше будет таких высших занятых обиталей. То есть, в большинстве реальных молекул ВЗМО – это не одна орбиталь, а целый пакет орбиталей. Иногда их нумеруют как-нибудь так ВЗМО, ВЗМО-1, ВЗМО-2 и т.п., или ВЗМО0, ВЗМО-1, ВЗМО-2 и т.п. На эти индексы смотреть не стоит: если орбитали расположены достаточно плотно, небольшие изменения в методике расчётов могут привести к изменению порядка.
Вот в этом пакете и ищем орбитали, которые соответствуют σ-связям в молекуле. Как это сделать, ведь молекулярные орбитали почти всегда многоцентровые? Во-первых, нужно чётко понять, какие из орбиталей относятся к π-системе, и не обращать на них внимания. π-Орбитали расположены всегда вне линий, соединяющих атомы, и вдоль этой линии электронная плотность равна нулю (линия лежит в узловой плоскости). Ищем именно σ-связи. Их легко найти – доли орбиталей в таких связях расположены вдоль линий, соединяющих атомы. Особенно нас должны будут заинтересовать орбитали, максимально локализованные на двух атомах связи (там будут наиболее жирные доли). Когда чёртов коронавирус оставит нас в покое, я сделаю несколько таких расчётов и выложу, чтобы это можно было увидеть и рассмотреть.
Наибоее часто такие локализованные ВЗМО будут у σ-связей углерода с более тяжёлыми элементами из 3,4,5-х периодов, и чем ниже, тем более локализованными будут связи. Это не только галогены, но и элементы 15 и 16 групп (фосфор и ниже, сера и ниже). Все такие связи более или менее легко подвергаются окислительному присоединению, хотя в реальности соотвествующие реакции не всегда легко найти, но они есть и в современной химии их уже много. Окислительное присоединение по связи C-P в лигандах мы обсуждали в теме Катализ, потому что это одна из побочных реакций, медленно, но верно, убивающих фосфиновые лиганды и сокращающая TON.
Связи с элементами 2 периода (фтор, кислород, азот) гораздо менее доступны. Соотвествующие им орбитали могут быть или вообще зарыты ниже пакета ВЗМО (это еще сильно зависит от того, что ещё висит с другой стороны на кислороде и азоте), или быть сильнее делокализованы. К тому же для большинства переходных металлов соотвествующие этим элементам лиганды – сомнительное приобретение. Поэтому, за исключением выдающихся уходящих групп типа трифлата, окислительное присоединение к таким связям до сих пор редкость и требует серьёзных усилий.
Нужно ли смотреть на энергию орбиталей, чтобы понять, насколько они дрступны для металла? Нет, это бессмысленное занятие. Нас интересует только положение – они должны быть в пакете ВЗМО. Энергия орбиталей в абсолютном измерении – вообще величина бесполезная, потому что это имеет мало отношения к реальности.
Какие выводы: Да, всё не безнадёжно, но в некоторой степени банально. Связи углерода с гетероатомами низних периодов действительно несложно найти в молекуле чисто визуально, но подбор металла и лигадов для того чтобы эти связи реально раскачать и запустить в какую-нибудь толковую реакцию остаётся на экспериментаторе. Второй вывод – использовать можно почти любую связь, но, возможно, только в направленном режиме.
Бухвальд
Ответ: Не только прощу, но и сам его так называю. И все остальные, кого я знаю, тоже. Как ещё мы можем прочитать эту явно немецкую (на самом деле, скорее идиш, восточноеврейский диалект, в основе имеющий немецкий язык) фамилию. Нам, советским людям, так и совсем тяжело. В советских газетах очень любили перепечатывать фельетоны некоего Арта Бухвальда, который на самом деле был вполне знаменитым левым журналистом в США во второй половине прошлого века. Он так славно бичевал язвы капитализма, при этом довольно остроумно, и с его писаниями сталкивался каждый советский человек. И так же как и наш Стефен, он тоже, по описаниям современников, дико удивился, когда однажды узнал, что его тексты не только тырят, не выплачивая гонорара, но ещё и так странно называют странные советские люди, потому что произношение его фамилии было, как и положено, Бакуолд (не Бачуолд). Поэтому здесь называйте как хотите, но когда отправитесь в США, не удивляйтесь, что вас не поймут.
Ибупрофен
Это такая разновидность процесса, используемого для синтеза уксусной кислоты BASF-Monsanto-BP. Спирт прямо в реакции превращается в галогенпроизводное. С бензильным это вообще легко происходит и можно взять просто HCl. Обычно гидроксикарбонилирование делают в присутствии оснований, но здесь это делают даже в присутствии небольшого количества кислоты, а низкие скорости стадий в таких условиях подстёгивают условиями пожёстче – в промышленном реакторе берут повыше температуру и давление. Ацильный комплекс расщепляется водой, а не гидроксид ионом.

Окислительное присоединение. Никель против палладия.
Ещё раз про окислительное присоединение. Во-первых, никакие формальные параметры типа электроотрицательности или тем более энергии орбиталей, не годятся для того, чтобы делать выводы о реакционной способности, тем более в конкретных реакциях. В химии вообще нет и не будет простых моделей, управляемых одним или хотя бы несколькими простыми параметрами. Всё, что нам дают такие вещи – весьма приблизительное обозначение общих тенденций.
И никель и палладий (и платина) в степени окисления 0 и конфигурации d10 обладают высокой способностью к окислительному присоединению. При этом, энергетика конкретной реакции зависит и от энергии разрываемой связи и от энергии образующихся связей на металле с двумя новыми σ-лигандами, углеродным остатком и бывшей уходящей группой. Именно в этом и состоит величие реакции окислительного присоединения по сравнению с нуклеофильным присоединением – уходящая группа, остающаяся в координационной сфере и в конечном продукте и в переходном состоянии, очень сильно снижает затраты на разрушение бывшей связи. В этом можно увидеть забавный парадокс – уходящая группа помогает разрушить связь, в которой она ещё недавно принимала участие. Поэтому мы и видим, как неожиданно полетели в самые разные реакции прежде весьма инертные с обычной органической химии вещества – ароматические и непредельные галогенпроизводные, трифлаты фенолов и енолов.
Никель отличается от палладия и тем, что это более маленький атом (кстати, не так уж и сильно, но заметно), а значит, он образует более короткие связи, особенно с элементами, более близкими к нему в Периодической системе. Он и предпочитает более маленькие атомы в лигандах, которые он связывает. По другому, это можно сформулировать так, что Ni(2+) является более жёсткой кислотой льюиса, чем Pd(2+). И никель поэтому получает более серьёзную компенсацию, связывая кислородные, азотные лиганды и даже фторид, да и хлорид тоже оказывается в преимущественном положении относительно других галогенов. Но здесь важно не впадать у сверхупрощение – важна и та связь, которая рвётся, и образующиеся. Поэтому с никелем больше шансов добиться активации в кросс-сочетании и других реакциях связей C-O, C-N, C-F, C-Cl.
При этом, не стоит забывать, что способность металла к окислительному присоединению можно сильно изменить с помощью анциллярных лигандов. И резервы палладия здесь даже больше, чем у никеля, как минимум потому, что у металла второго ряда больше места в координационной сфере, и там можно разместить более серьёзные лиганды. С эти связаны большие успехи в химии палладия в вовлечении хлорпроизводных, а такэе некоторых менее активных сульфонатов (тозилатов, мезилатов). С другой стороны, потолок у палладия всё равно ниже, чем у никеля.
Бета-гидридное элиминирование
Сразу начну с конца. Нет, не может быть процесс устроен так, что образующийся при гидридном элиминировании алкен сразу уходит из координационной сферы, освобождая место. В этом смысл всей химии переходных металлов – каждая реакция происходит в координационной сфере, а выход из неё – отдельный процесс. И ещё не нужно забывать, что большиснтво процессов в этой химии обратимо, и прямой процесс должен соотвествовать обратному. Реакция миграционного внедрения – β-гидридного элиминирования. Олефин по определению на металле. Если заблокирована одна реакция, заблокирована и другая.
Теперь про блокирование элиминирования дифосфинами и о том, необходимо это или только достаточно или наоборот.
Увы, в химии так не бывает. В химии бывает по-другому. Встает задача какая-то, напрмер, проблема изомеризации алкила в координационной сфере. Большие учёные чешут репы и выдвигают мощную идею. В первом же опыте идея работает. Опыт входит в историю, становится классикой, придумавший его большой учёный становится еще больше, все ему или ей поклоняются, как живому божеству. Но последующие повторные использования идеи почему-то работают без того блеска, или вовсе не работают. Пеняет ли кто-нибудь живому классику, что что-то пошло не так? Ну нет конечно. Не принято. Иначе в науке авторитетов не останется, а они всё же нужны. Тем более, что обычно идея всё таки иногда работает, и еще пара-тройка впечатляющих примеров находятся.
Вот эта история с подавлением гидридного элиминирования ровно из этого ряда. Исследование про то, как можно сделать кросс-сочетание с алкильным субстратом и подавить изомеризацию с помощью новомодного лиганда dppf произвела в те времена просто ошеломляющее впечатление. Это точно стало классикой этой науки. Но и что – стали алкильные субстраты с тех пор столь же распространены в кросс-сочетании, как и арильные? Нет. Потому что до сих пор изомеризации остаются проблемой, а с алкильными субстратами обычно не хотят связываться. А что же dppf? Иногда работает, но чаще нет. Да и другие бидентатные лиганды тоже.
Но сказать, что это совсем не работает невозможно. Та же идея как минимум ещё один раз здорово сыграла, когда Хартвиг и Бухвальд порознь смогли подавить гидридное элиминирование из аминов, и тем же dppf и BINAP’ом. Но и здесь это работает не на сто процентов, и бывают досадные проколы.
В этом вся суть химии. Ни один приём не работает везде и всегда, а красивое объяснение, возможно, высосано из пальца (но при этом работает, хотя и не всегда). Это потому что запрет в химии это не запрет в строгом смысле, а выбор между менее и более выгодным, и как всякий такой выбор, он может сработать, а может и не сработать. Почему может не сработать бидентатный лиганд? Минимум по двум причинам. Мы считаем его координационно стабильным, а химию палладия – химией координационного числа 4. Но, во-первых, стабильный лиганд может оказаться в каких-то условиях гемилабильным, да и образование 5-координированных комплексов для палладия не запрещено. Надо понимать, что обы эти пути менее выгодны, чем обычный, тот самый, который и объясняет нам запрет. Но менее выгодны не значит невозможны. Они могут быть побочными путями, снижающими селективность, и так обычно и бывает. То есть, здесь выбор не качественный (да/нет, всем сидеть дома), а количественный (а можно мне? выйти с собакой на 100 метров, ой, а получилось на 10 километров, и без собаки).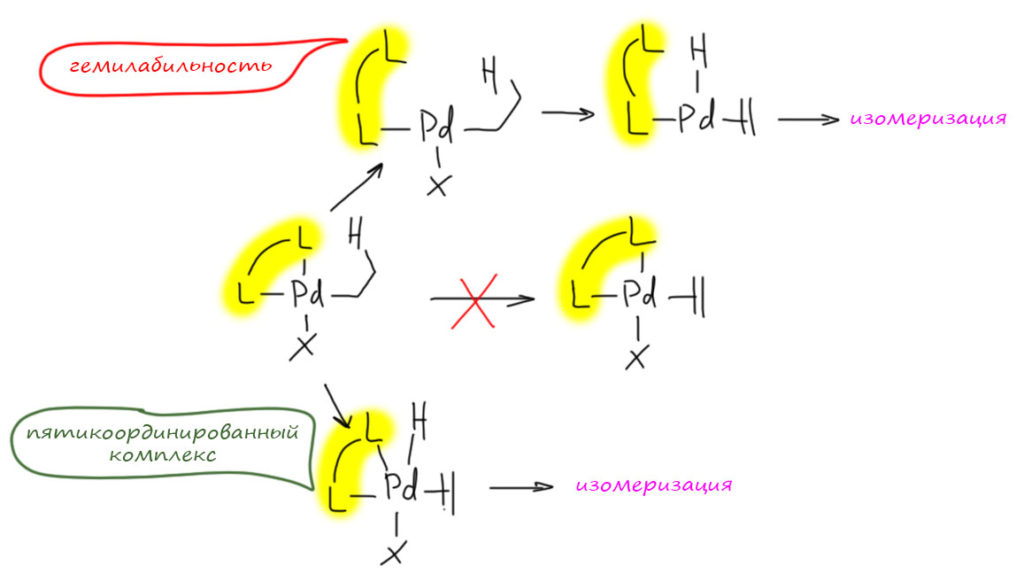
А что же с этим делать, если заранее точно нельзя сказать? А то же самое, что всегда делали химики – ставить реакции, пробовать, и иногда добиваться успеха.
Реакция Соногасиры без меди
Совершенно верно. Реакция Соногасиры стала именно реакцией Соногасиры (то есть с солью меди(I) в присутствии третичного амина) именно потому что она очень надёжна и дает отличные результаты. Невообразимое количество всяких ацетиленов получено с ее помощью. Это точно одна из самых популярных реакций с участием комплексов переходных металлов, уступающая только реакции Судзуки, но точно сильно превосходящая все другие методы кросс-сочетания.
Реакция без меди гораздо менее надёжна (ее кстати иногда называют реакцией Соногасиры-Хека потому что условия – простой комплекс палладия и третичный амин – практически идентичны самым распространеннным условиям реакции Хека, открытые Хеком). Почему? По разным причинам, но в основном потому что она менее селективна. Вообще-то в таких условиях скорее должно происходить карбопалладирование с образованием аддукта, которому некуда дальше превращаться (водород у него с другой стороны), и пока разбирались в этих вещах, много времени ушло.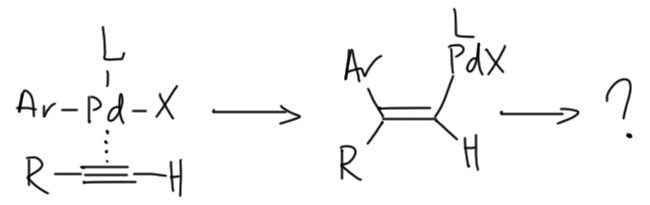
Но иногда вместо этого идёт то же, что в обычной Соногасире. Это означает, что из координированного ацетилена основание может отнять протон, и тогда получится то же самое. Это конкурентный путь. 
В начале развития этой химии не нашли хороших условий, когда этот путь становится основным, а за это время Соногасира с медью ушла так далеко вперёд, и решила так много задач, что мало кто задумывался об альтернативе.
Но времена меняются и к Соногасире-без-меди вернулись. И в последние 10 лет она становится всё более популярной. Почему? Во-первых, потому что у Соногасиры-с-медью есть большая проблема – она применима к очень узкому кругу электрофилов, в основном к иодпроизводным и немного к бромпроизводным, и то со скрипом. А современная химия уже почувтствовала вкус к широкому кругу уходящих групп. А Соногасира-с-медью очень плохо поддается на варьирование анциллярных лигандов – это очень консервативная реакция. И когда стали пытаться варьировать лиганды, убедились, что довольно часто приходят к возможности убрать медь из протокола.
Во-вторых, современная наука, как мы знаем, испытывает колоссальное давление со стороны шведского малолетнего чудовища. И стало просто необходимо всё делать максимально зелёным, иначе придёт Грета и мало не покажется. Медь надо убирать точно – это плохой элемент, он загрязняет среду. И когда Соногасира используется для синтеза, например, лекарств, очистка продукта от меди – страшно затратная процедура, а с остаточной медью лекарство на рынок не выйдет, тут даже Грета ни при чём. А меди в Соногасире-с-медью, как назло, кладут очень много, намного больше палладия. И если палладий можно сильно сократить с помощью новомодных лигандов, то с медью это не получается. Вот и начали опять исследовать Соногасиру-без-меди (это называют именно так, хотя стоило бы вспомнить тех, кто это сделал раньше, ведь, по крайней мере, Луиджи Кассар ещё жив, правда занимается какой-то странной фигнёй, но очень-очень экологически правильной, угодной Грете). Есть и очень серьёзные достижения. Но в битве Соногасиры-с-медью с Соногасирой-без-меди пока не будет победителей, потому что в реальных синтезах приходится пробовать и то, и это, и в одних даёт лучшие результаты первая, в других вторая. Пока между ними заключено водяное перемирие.
Реакция Хека и кросс-сочетание
Этот вопрос можно было бы счесть неактуальным, но в нём есть смысл, и я коротко ещё раз на этом остановлюсь. Реакция Хека (Мидзороки-Хека) – это кросс-сочетание или нет?
С формальной точки зрения это несомненно кросс-сочетание. Ведь мы связываем два разных фрагмента простой связью. Более того, это ещё и CH-активация, ведь со стороны олефина разываеся именно CH-связь.
Более того, Ричард Хек стал лауреатом Нобелевской премии вместе с Акирой Судзуки и Эй-Ити Негиси, и эту премию принято называть “нобелевкой за кросс”, и поэтому иногда считают, что именно его пристегнули к двум японцам, настоящим отцам кросс-сочетания, типа потому что нобелевка без американца вызовет отправку авианосца “Джон Ф Кеннеди” к берегам Стокгольма. Но это совсем не так. Именно Хек в этой нобелевке главный и безо всякого авианосца. Хеку принадлежит, наверное, самый впечатляющий вклад в реакции с участием переходных металлов на самом первом ее этапе, и это и карбонилирование, и гидроформилирование, и кросс-сочетание, и реакции олефинов. Японцы, и не только эти, безусловно, мощно потрудились, чтобы создать эту науку, но работали они уже на основе, заложенной Хеком. Поэтому эта нобелевка совсем не говорит о том, что реакция Хека это кросс-сочетание, она говорит, что без Хека кросс-сочетания могло бы и не быть.
А реакция Хека – это не кросс-сочетание. Кросс-сочетание принято понимать не только как соединение фрагментов простой связью, но несколько более узко – как реакцию, в которой участвуют (разрываются и образуются), в том числе и на стадиях механизма (каталитического цикла, если оно каталитическое) только σ-связи. Было два реагента с простыми связями, и получилось два продукта, и в них простые связи переехали на новые места. И это не только так, если сравнивать исходные и конечные, но и по дороге. Поэтому в кросс-сочетаниях участвуют только реакции, имеющие дело с простыми связями – окислительное присоединение, восстановительное элиминирование, лигандный обмен (замещение), переметаллирование, металлирование, метатезис σ-связей со всеми своими разновидностями. Но не участвуют реакции с участием кратных связей двойных, тройных, аллильных систем и т.п. Кстати, поэтому не только Хек, но и аллильное замещение не является кросс-сочетанием.
Но и последняя ложка дёгтя в бочке битума. Кто это сказал? Государь всея химии? Начальник мирового правительства? Главный сионский мудрец? Нет, никто, нет таких персонажей, по крайней мере, в химии. Просто так принято. Не хотите, не соглашайтесь. Имеете право. В серьёзной науке никаких верховных авторитетов нет, и если вам кажется важным считать по-другому, и если вы сможете убедить в этом хоть кого-то, то вперёд.
Аддукт окислительного присоединения - стереохимия
Вы правы, но это не важно. Почему не важно? А потому что это ни на что не влияет. Вообще в любом превращении, в любом механизме всегда очень много деталей, но не все они заслуживают рассмотрения или даже упоминания, но только те, которые влияют на ход реакции. В самом начале этому уделили внимание, потому что a priori кажется, что это должно быть важно. Уж точно, к восстановительному элиминированию комплекс должен подойти с цис-расположением нужных лигандов, иначе не будет никакого восстановительного элиминирования. И кажется, что все нормально, потому что аддуктом окислительного присоединения должен быть цис-аддукт. А переметаллирование вроде должно идти с сохранением.
Но вот засада. Аддукты окислительного присоединения очень часто вполне стабильны и легко выделяются. И структур таких аддуктов нарисовали множество уже в самом начале этой химии. И, как назло, почти все они имели транс-конфигурацию. Именно такие комплексы выделяются из растворов, кристаллизуются. Довольно понятно почему – они симметричнее цисов, а симметричные молекулы всегда легче пакуются в кристалл. ![]()
Но это значит, что есть изомеризация, и она, вероятно, обратима. Это не такая простая мысль потому что даже маленьким детям известно, что плоскоквадратные комплексы металлов 10 группы конфигурационно устойчивы, и легко выделяются в цис- и транс-формах. Про цисплатин и трансплатин слышали все. Ну да, только в химии никогда не бывает абсолютных правил. В этих классических комплексах с маленькими лигандами всё так, а в аддуктах окислительного присоединения не так. Транс-форма не только легко выделяется (это ведь прямо не связано с устойчивостью, а только с кристаллизуемостью), но и правда более устойчива. Или по стерическим причинам – даже простой трифенилфосфин это немаленькая нашлёпка на шарике палладия. Возможен, и пресловутый транс-эффект или другие эффекты, связанные с тем, что противоположные лиганды сидят на одной многоцентровой орбитали. Нам это не важно.
Как изомеризуются такие комплексы? Или диссоциативно-ассоциативно или наоборот. Оба пути возможны, потому что такие комплексы 16-электронные и могут и туда, и сюда. Пятикоординированные комплексы координационно неустойчивы из-за хорошо известного конформационного превращения, называемого псевдовращением. Трёхкоординированные – еще проще.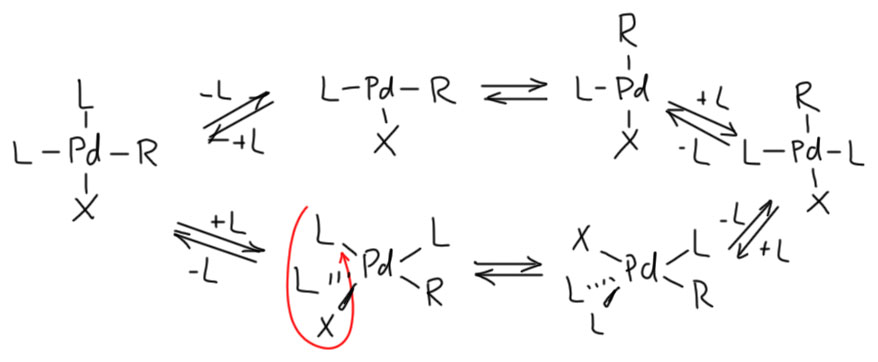
Вообще-то нам это до лампочки. Мы ведь занимаемся переходными металлами только для органической химии, а не координационной химией как таковой. Все, что имеет значение для орагники нас волнует, все остальное – только если останется время на это.
И конечно, хелатные комплексы, а также комплексы с монодентатными лигандами типа модных фосфинов Бухвальда, которые эффективно экранируют второе координационное место, формально свободное, своими огромными тушами дают устойчивые цис-аддукты. С такими лигандами как КсантФос вообще все еще сложнее. Это хорошо, потому что цис-транс-изомеризация, если она есть, безусловно отнимает у скорости каталитического цикла. Но – и это страшный секрет всей нашей химии – все эти кросс-сочетания и прочее на деле реакции весьма небыстрые – посмотрите на типичные TOFы и вы почти никогда не увидите такой единицы измерения как обратные секунды. А вот обратные часы и даже обратные дни не будут редкостью. В такие времена не то что изомеризацию, а даже и небольшую революцию вписать можно – никто и не заметит.
Реакция Бухвальда-Хартвига - с чего начинается цикл?
Имеется в виду вот эта схема. 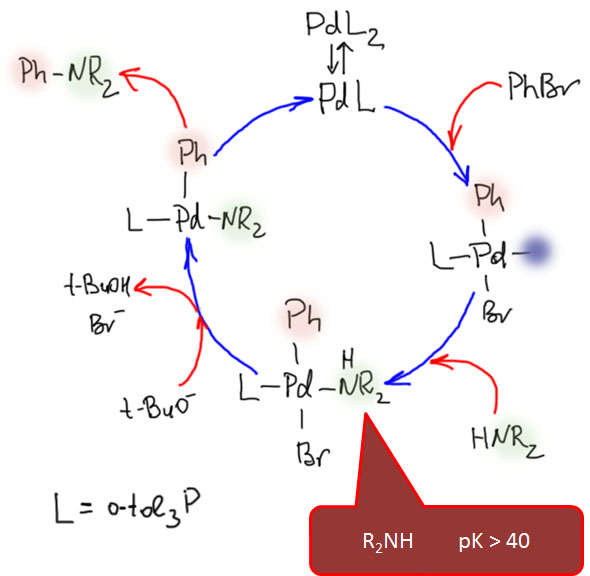 Во-первых, еще раз повторю, в реакцию вступает пред-катализатор. В простых случаях это комплекс палладия с 4 или 3 лигандами типа триарилфосфина. В реакциoнной смеси такой комплекс находится в равновесии с комплексами с меньшим числом лигандов. Это ясно говорит нам координационная химия – у каждого комплекса есть константы устойчивости, которые собственно и описывают эти равновесия. Чем меньше лигандов остается, тем меньше константа. Но, особенно при повышенной температуре в растворе присутствуют все комплексы и даже некоторое количество “пустого” палладия (на самом деле там просто молекулы растворителя вместо фосфинов). И положения этих равновесий ещё зависят от того, какой комплекс вы берете как предкатализатор. Если обычный трифенилфосфиновый, то вы начинаете с PdL4 и поэтому у вас много комплекса с тремя лигандами, мало, но ощутимо комплекса с двумя и очень-очень мало комплекса с одним. Если у вас комплекс с тритолилфосфином, который более объёмист, то на палладии сразу помещается только три таких лиганда, поэтому комплекса с одним уже побольше – до него уже рукой подать.
Во-первых, еще раз повторю, в реакцию вступает пред-катализатор. В простых случаях это комплекс палладия с 4 или 3 лигандами типа триарилфосфина. В реакциoнной смеси такой комплекс находится в равновесии с комплексами с меньшим числом лигандов. Это ясно говорит нам координационная химия – у каждого комплекса есть константы устойчивости, которые собственно и описывают эти равновесия. Чем меньше лигандов остается, тем меньше константа. Но, особенно при повышенной температуре в растворе присутствуют все комплексы и даже некоторое количество “пустого” палладия (на самом деле там просто молекулы растворителя вместо фосфинов). И положения этих равновесий ещё зависят от того, какой комплекс вы берете как предкатализатор. Если обычный трифенилфосфиновый, то вы начинаете с PdL4 и поэтому у вас много комплекса с тремя лигандами, мало, но ощутимо комплекса с двумя и очень-очень мало комплекса с одним. Если у вас комплекс с тритолилфосфином, который более объёмист, то на палладии сразу помещается только три таких лиганда, поэтому комплекса с одним уже побольше – до него уже рукой подать.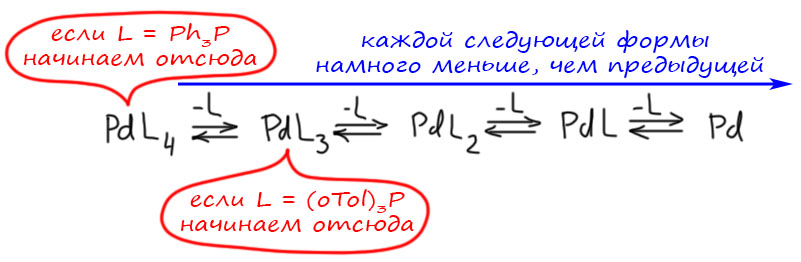
И теперь мы решаем замутить каталитический цикл. У вас все формы комплексов в растворе, но цикл предъявляет запрос только на какой-то конкретный, потому что по дороге требуется столько-то свободных мест. Вот и самый первый цикл реакции C-N кросс-сочетания требовал три места, чтобы разместить там два куска галогенпроизводного и амин. Поэтому такой цикл отбирает монолигандный комплекс. До времени пустующее место (на самом деле занятое растворителем) на схеме показано фиолетовым пятном – там неявный лиганд.
В принципе, мы могли бы начинать с комплекса PdL2 и ожидать диссоциации лишнего лиганда уже после окислительного присоединения. Ничего бы в общей схеме это не изменило, а такого рода детали реально выяснить практически невозможно, да это и не важно, никто время тратить не будет на такое исследование, даже если бы его можно было сделать.
Краткий курс истории КсантФоса
“В чём именно механизм…” Такие вопросы отражают немного наивное желание видеть простые и точные объяснения для очень сложных явлений. Мы уже не первый раз с этим сталкиваемся. Но так не бывает. Еще раз повторю – все процессы в химии очень сложны и зависят от множества факторов, которые еще и влияют друг на друга. В химии нет линейных эффектов. Очень модная в прошлом веке теория линейных зависимостей между реакционной способностью и структурой (это называлось LFER – linear free energy relationships) в общем в прошлом веке и осталась. Тогда писали серьёзные книжки под названиями что-то типа Основы количественной теории реакционной способности, и всерьёз намеревались сделать органическую химию почти точной наукой. Но химия – не точная наука, химия – естественная наука, она основана не на конструировании объектов, а на наблюдении над реальными объектами. И всё это поэтому недалеко ушло от первого и действительно фундаментального достижения этой теории, уравнения Гаммета, хотя пыталось уйти не просто далеко, а очень далеко. Химия, что органическая, что любая другая, та её часть, которая имеет дело с реакциями и реакционной спсобностью, упорно и бешено сопротивляется всему, что выходит за рамки простых обобщений и грубых качественных тенденций.
Вот и в той науке, что мы изучаем, последовательность действий всегда одна и та же: исследователи сталкиваются с проблемой, в попытках ее решения выдвигают гипотезы, пытаются создать нечто, удовлетворяющее этим гипотезам, иногда достигают успеха, и провозглашают гипотезу работающим методом. В этом месте обычно возникает ажиотаж, создателей гипотезы прославляют как крупных учёных (справедливо, они таковы и есть), воздают им почести (тоже правильно, кому же ещё их воздавать), и множество других, более скромных учёных бросаются воплощать метод в жизнь. И всегда происходит одно и то же: метод иногда работает, иногда нет. Что из этого следует? Только то, что гипотеза была приближением к реальности, схватила что-то очень важное, а остальным закономерно пренебрегла. Пользоваться можно? Можно. Но ожидать того, что раз и навсегда открыт ключ ко всему подобному не стоит.
КсантФос – очень яркая иллюстрация к этому. Есть смысл немного подробнее рассказать историю того, как он появился в этой науке, чтобы увидеть, как много здесь определяется не рациональным расчётом, основанным на точном понимании механизма и других умных вещей, а случаем, везением и настойчивостью.
Этот лиганд был придуман вместе с целой серией похожих лигандов мощным голландским исследователем Питом ван Лиуеном (Piet W. N. M. van Leeuwen et al. Organometallics 1995, 14, 3081-3089) для того чтобы добиться максимальной региоселективности в гидроформилировании пропилена.
Отсылаю на слайд про гидроформилирование в лекции про CO – задача в том, чтобы увеличить выход нормального альдегида и максимально подавить образование альдегида изо-строения. Для этого сначала от кобальтовых катализаторов перешли к родиевым, затем на родий стали вешать фосфины и искать такие фосфины, которые дают максимальеую селективность. Очевидно, что здесь должна работать стерика, но этого мало. Больший эффект дают дифосфины с большим углом укуса. Стали искать такие дифосфины, пришли к молекулам типа DPEPhos, но эффет был невелик. Здесь надо сказать, что уже с трифенилфосфином соотношение нормального к изо составляет 9:1. Но для промышленности это плохо. Эффект более крупных дифосфинов таков что выход изо снижается до 2-3%. И именно XantPhos даёт лучший результат, снижая выход изо до менее 1%.
Внимание, вопрос. Как сам ван Лиуен, один из крупнейших специалистов по катализу комплексами переходных металлов, объясняет свой результат? Учёный, с удовольствием нахваливая свой новый лиганд, говорит, что они экспериментально выяснили, что селективность увеличивается с увеличением угла укуса вплоть до значений, когда хелатирование становится невозможным. И что жесткая структура лиганда, вероятно, способствует особой устойчивости хелатов, в том числе при повышенной температуре. Вот так, чисто приблизительно, и где тут тонкости работы катализатора. И кстати, в дальнейшей истории этого лиганда было довольно ясно показано, что объяснения ван Лиуена были неверны, и хелаты КсантФоса вовсе не так стабильны, как ему казалось, и очень даже возможно, что именно в этом его сила, – но и это не вполне достоверно.
Такой потрясающий лиганд должен отлично работать и в других сложных случаях. Ван Лиуен считал, что он отлично держит координационную сферу, он объёмист и имеет один из самых больших углов укуса. Хорошая реакция, где это тоже должно работать – кросс-сочетение с алкильным Гриньяром. Помните классику жанра – dppf подавляет изомеризацию. КсантФос должен быть ещё лучше. Сам же ван Лиуен и организовал исследование (Eur. J. Inorg. Chem. 1998, 155). И КсантФос позорно провалился. А вот DPEPhos сработал даже чуть лучше, чем dppf.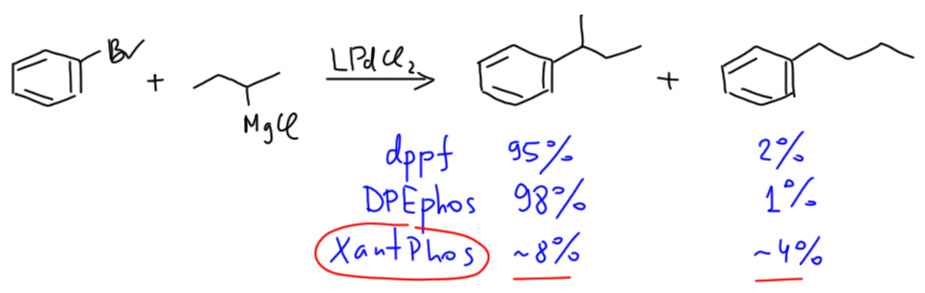
Другой тест был на аллильном замещении. Задача была в том, чтобы селективно получить менее замещённый продукт замещения. Попробовали целую кучу дифосфинов от обычных dppe, dppp, dppb до dppf и лигандов ван Лиуена. В общем-то неплохо сработали все, даже с самым плохим dppe выход желаемого продукта был 96%. C dppf получилось лучше, а с DPEphos совсем хорошо. Ксантфос тоже не провалился, но реакция была в пять раз медленнее. Очевидно, что избыточный стерический объём не всегда помогает.
В общем, после первого триумфа последующие шаги ксантфоса были не очень убедительны. Рядом с ним DPEPhos, первоначально забракованный ван Лиуеном, просто сиял. Неплохо DPEPhos себя показал в реакции Бухвальда-Хартвига в кросс-сочетании анилинов с бромбензолами (Tetrahedron Lett. 1998, 39, 5327). Впрочем, сам ван Лиуен показал, что КсантФос всё же неплохо работает в C-N кросс-сочетании и алкиламинов, и вторичных аминов с бромпроизводными, но никаких особенных преимуществ по сравнению с уже ставшим классикой протоколом Бухвальда с BINAP в нем не было.
В новое столетие мы вступили с очень простыми взглядами – все понимают, что для тех случаев, когда есть проблемы в восстановительном элиминировании, желательно использовать хелатирующие дифосфины с большим углом укуса. Но насколько он должен быть велик, и можно ли прямо точно выбрать лиганд для конкретной задачи, – на это ответов не было. Все по-прежнему решалось перебором, но кандидаты для перебора брались из понятного множества. И над всем этим хаосом носится ван Лиуен, очень влиятельный и уважаемый человек в катализе, он всем рассказывает про свои фосфины, дарит образцы и всех побуждает пробовать, особенно тем, кто упёрся в восстановительное элиминирование. А надо сказать, что самой главной реакцией в это время было аминирование – открытие Бухвальда и Хартвига свело все с ума, все хотели что-то достичь в этой области, найти способ применить реакцию к другому типу NH-нуклеофилов, а разных NH-нуклеофилов невероятно много и первоначальные достижения в этой области затронули практически только разные амины, и то, в основном достаточно простые. Своей очереди ожидали гетероциклы, амиды, гидразины, азид, и т.д.,а за почти за каждым из этих слов скрываются не отдельные соединения, а ряды рядов. И мы знаем, что в C-N кросс-сочетании восстановительное элиминирование всегда является проблемой, но с простыми NH-нуклеофилами ее решили бинапом и dppf, но с более хитрыми всё упёрлось. Все взалкали новых лигандов.
И вот, прямо на рубеже веков и тысячелетий, практически одновременно появляются две работы. Алексей Сергеев под руководством Галины Артамкиной и Ирины Белецкой сделал арилирование мочевины бромбензолами (Artamkina, G. A.; Sergeev, A. G.; Beletskaya, I. P. Tetrahedron Lett. 2001, 42, 4381). Мочевина – амид угольной кислоты, даже ещё более ленивый нуклеофил, чем просто амиды. Ключом к решению оказался КсантФос. 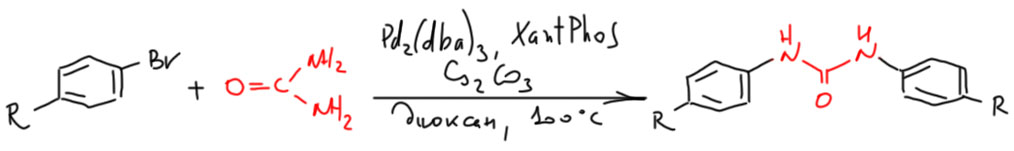
А Йин и Бухвальд опубликовали протокол арилирования первичных амидов и сульфонамидов сначала в кратком сообщении (Yin, J.; Buchwald, S. L. Org. Lett. 2000, 2, 1101), а затем в огромной статье с множеством реакций и исследованием комплексообразования КсантФоса (J. Am. Chem. Soc. 2002, 124, 6043). Тот же Бухвальд и обнаружил, что при окислительном присоединении КсантФос образует аддукт с транс-конфигурацией (точно так же как обычные фосфиновые комплексы – мы уже обсуждали, что в этих случаях почти всегда выделяются комплексы с аткой конфигурацией). ![]() Это открытие озадачило основоположника, так как ему пришлось как-то объяснять, как вообще такой комплекс может участвоать в дальнейшем ходе каталитического цикла. Надо сказать, что Бухвальд невероятно крут, когда нужно что-то придумать, и столь же невнятен, когда нужно что-то объяснить. Читать статьи Бухвальда – тяжкий труд и адская головная боль. Вот и тут раздалось нечто невразумительное. Рано или поздно понадобится цис-конфигурация, иначе не видать нам восстановительного элиминирования. В общем, главным выводом на этот момент стало признание того, что координационная химия у КсантФоса, мягко говоря, сложнее, чем предполагалось, а уж как это связано с выдающимися свойствами этого лиганда в арилировании амидов и мочевины, пока что осталось еще более загадочным, чем было до этой работы.
Это открытие озадачило основоположника, так как ему пришлось как-то объяснять, как вообще такой комплекс может участвоать в дальнейшем ходе каталитического цикла. Надо сказать, что Бухвальд невероятно крут, когда нужно что-то придумать, и столь же невнятен, когда нужно что-то объяснить. Читать статьи Бухвальда – тяжкий труд и адская головная боль. Вот и тут раздалось нечто невразумительное. Рано или поздно понадобится цис-конфигурация, иначе не видать нам восстановительного элиминирования. В общем, главным выводом на этот момент стало признание того, что координационная химия у КсантФоса, мягко говоря, сложнее, чем предполагалось, а уж как это связано с выдающимися свойствами этого лиганда в арилировании амидов и мочевины, пока что осталось еще более загадочным, чем было до этой работы.
Спустя почти пять лет свой вклад внёс Хартвиг. Надо сказать, что до арилирования амидов Бухвальд и Хартвиг бежали по дорожке как два образцовых чемпиона – даже фотофиниш не мог бы прояснить, кто из них раньше преодолевал очередной этап странной эстафеты, в которой все этапы бегут одни и те же. Но именно на арилировании амидов Хартвиг отстал – нет у него альтернативной Бухвальду работы на эту тему. Расслабился, а скорее увлёкся другой химией – в самом конце самой последней лекции даже станет понятно какой. Зато потом отомстил. Бухвальд в работе 2002 года сетует, что они не смогли получить амидный комплекс ксантфоса и подобраться поближе к тому месту, где в каталитическом цикле скрыт эффект этого лиганда. А Хартвиг смог, и даже рентген сделал (не с обычным амидом, а с сульфонамидом, но это не важно, J. Am. Chem. Soc. 2006, 128, 9044). И увидел Хартвиг, что это тоже во-первых, транс-комлекс (вернусь на химфак, сделаю сюда картинку этого комплекса). В этом месте можно было бы не радоваться, а горько заплакать, потому что чёрт его знает, как этот транс-комплекс может объяснить хоть что-то. Но не на того напали. В химии важно не просто получить интересный результат, но гораздо важнее получить данные для хорошего сравнения, потому что в химии всё познаётся не само по себе, а именно в сравнении. В химии важно не состояние, которое всегда куда-то ускользает, а хорошая тенденция – мы не знаем, как эта штука работает, но немного понимаем, чем она отличается от других. Совсем немного понимаем, но хоть так. Один транс-комплекс ксантфоса можно было бы засунуть … в кембриджскую базу данных. Но Хартвиг получил и сделал структуры еще для нескольких амидных комплексов, но с монодентатными лигандами. И увидел, что в них амид связан как хелат, азотом и кислородом. 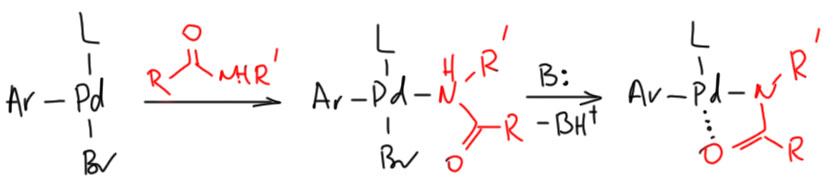 И еще он показал, что КсантФосный комплекс, хоть и транс-, но при длительном нагревании приводит к восстановительному элиминированию, а комплекс с хелатным амидом в тех же условиях – не приводит (эх, ну, точнее, приводит, но за 30 часов против пяти). Отсюда и пошла теория, что КсантФос не даёт амиду давать хелатный комплекс.
И еще он показал, что КсантФосный комплекс, хоть и транс-, но при длительном нагревании приводит к восстановительному элиминированию, а комплекс с хелатным амидом в тех же условиях – не приводит (эх, ну, точнее, приводит, но за 30 часов против пяти). Отсюда и пошла теория, что КсантФос не даёт амиду давать хелатный комплекс.
Оцените, на каком замечательном основании стоит эта теория. Я бы назвал это тонким льдом над трясиной. Во-первых, совершенно очевидно, что точно так же должны вести себя любые дифосфины, хоть тот же dppf или BINAP. Но – не работают они в арилировании амидов. Была еще такая идея, что в комплексах КсантФоса как-то участвует атом кислорода, который на обычной картинке как будто находится прямо под атомом металла (и, видимо, щекочет ему брюшко своими неподелёнными парами, отчего тот становится нервным и быстрее выпихивает висящие на нём лиганды). Увы, это не подтверждается ни сравнением с другими похожими лигандами, ни реальной структурой комплексов, которые не являются плоскими (в лекции на слайде есть рентгены – там это видно). Во-вторых, так как же транс-комплекс даёт восстановительное элиминирование?
Приходится признать, что хелаты КсантФоса должны проявлять гемилабильность. Другого пути превратить транс-комплекс в цис-, а без него нет восстановительного элиминирования, не придумать. Гемилабильность КсантФоса по всем признакам проявляется только в достаточно жестких условиях и скорость этого процесса весьма мала – это, кстати, даем весьма невысокие и TON, и особенно TOF в таких реакциях, но другого решения все равно нет. Во время цис-транс изомеризации амид очевидно не успевает зацепиться за палладий кислородом (это, в любом случае более слабая связь чем Pd-P, и термодинамика все равно выиграет). В цис-комплексе КсантФос имеет очень большой угол укуса, а значит комплекс очень напряжён и это помогает “выпихиванию” продукта. Объяснение особой роли КсантФоса, на мой взгляд, может быть в том, что после выпихивания комплекс возвращается в более выгодную и менее напряжённую транс-конфигурацию. Именно поэтому он так хорошо и работает в тех случаях, когда нужно как-то подтолкнуть тяжелое восстановительное элиминирование. 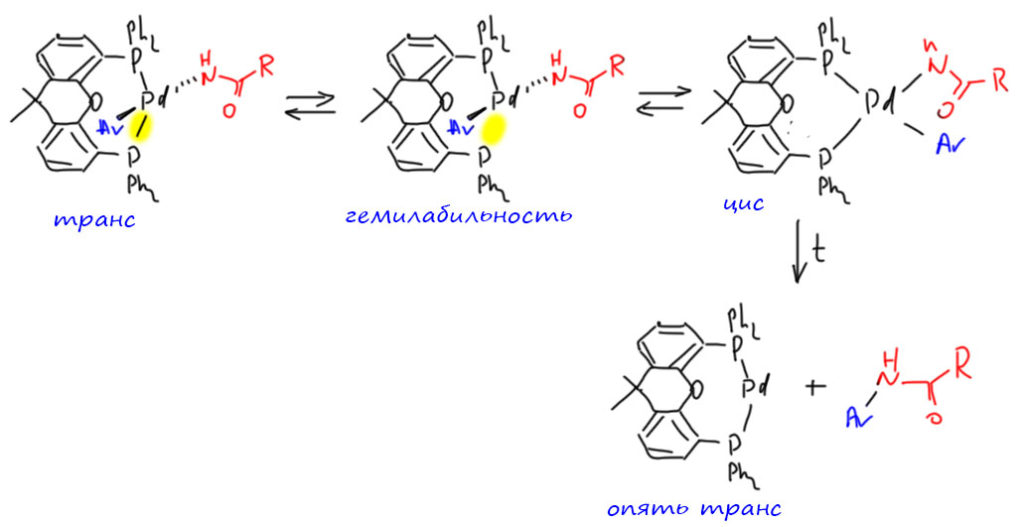
Подводя некоторый итог скажем, что особые свойства КсантФоса, видимо, в уникальном сочетании нескольких свойств:
- он может образовывать как цис- так и транс-хелаты;
- транс-хелаты более устойчивы и поэтому весьма легко образуются, в них палладий проводит большую часть каталитического цикла
- высокая устойчивость транс-хелатов делает каталитические системы устойчивыми и долгоживущими (этим свойством КсантФос’ного катализа восхищался ещё ван Лиуен)
- цис-хелаты менее устойчивы и сильно напряжены из-за большого угла укуса, изомеризация из транс в цис происходит при повышенной тмпературе и довольно медленно
- в цис-хелатах легко происходит восстановительное элиминирование, что приводит, среди прочего к ослаблению напряжения в хелатном цикле
В последние 15 лет КсантФос стал почти незаменимым лигандом в кросс-сочетании и других реакциях. Говорят, что по встречаемости в реакциях он уступает только BINAP’у. Во всех случаях, когда есть проблемы с восстановительным элиминированием (оно или вообще не происходит с другими лигандами, или происходит, но нужно ускорить, чтобы избежать побочных реакций) пробуют КсантФос. Вот только один пример, сразу две группы, во Франции (S.Perrio et al. Tetrahedron 2005, 61, 5253) и Японии (T. Itoh, T. Mase Org. Lett. 2004, 6, 24, 4587-4590) публикуют очень похожие протоколы C-S кросс-сочетания, в которых КсантФос опередил все остальные лиганды. В C-S кроссе восстановительное элиминирование тоже идет не очень весело, и КсантФос ему в помощь.
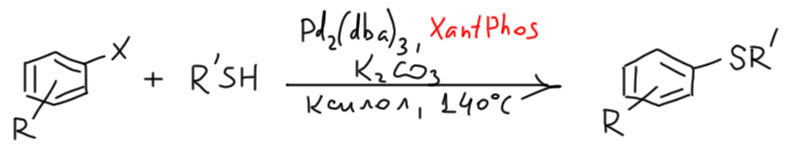
К сожалению или к счастью, химия устроена так, что ничто не работает как швейцарские часы, и КсантФос не исключение. Но во многих случаях это просто незаменимый лиганд. А представьте, что ван Лиуен его не придумал бы? В этой молекуле нет ничего такого, что говорило бы, что её обязательно нужно сделать и попробовать. А если бы ван Лиуен не был так настойчив и не совал бы его всякому встречному и поперечному на всех конференциях? Будьте как этот великий голландец, если получили что-то стоящее, верьте и продвигайте. На лежащий в банке в темном углу тяги лиганд палладий сам не запрыгнет.
А что же с хелатным связыванием амида? Это не так важно? Да нет, это серьёзный фактор, и Хартвигу можно сказать большое спасибо, что он его обнаружил и разоблачил. В нескольких очень свежих работах было показано, что на скорость реакции с амидами можно влиять и другим способом – добавлением кислот Льюиса, роль которых, веротяно, сводится к координированию амидного лиганда по кислороду, что тоже не даёт ему зацепиться хелатным способом за палладий. Пока оставим эту историю.
В завершение краткой истории триумфального шествия КсантФоса скажем, что на фосфоре там два фенила. А следовательно от этого лиганда невозможно ожидать, что он, например, сможет способствовать окислительному присоединению к менее реакционноспособным связям, например, к C-Cl. И очевидная идея вместо фенила повесить туда циклогексилы или трет-бутилы не работает. Почему? Тонкая это штука, равновесия комплексообразования, от которых зависит тонкая настройка каталитических циклов. Заменили фенилы – и сломали. А если мы хотим арилировать амиды хлорпроизводными? В наше время это уже стало хорошим тоном. Ищите новый лиганд. Уже не надо. Его нашёл Бухвальд. С помощью постдока по имени Brett Fors. Лиганд назвали BrettPhos (оцените игру слов – так и видишь, как Бухвальд, заполучив такого чувака, отправил его в лабораторию с напутствием: Иди работай, и без нового выдающегося лиганда не возвращайся, имя ему уже есть – и чувак не подвёл). Это типичный для Бухвальда монофосфин с затейливой формой молекулы, которая защищает второе место на палладии, формально не занимая его координацией. Вся остальная обвеска молекулы осуществляет тонкую настройку. Лиганд существует, как всегда у Бухвальда в двух формах, основной с циклогексилами, и еще более рогатой с трет-бутилами. Понятно, что такая штука и арилхлорид присоединит, и амиду не даст распоясаться, и продукт с треском выпихнет (Tetrahedron 2009, 65, 6576). 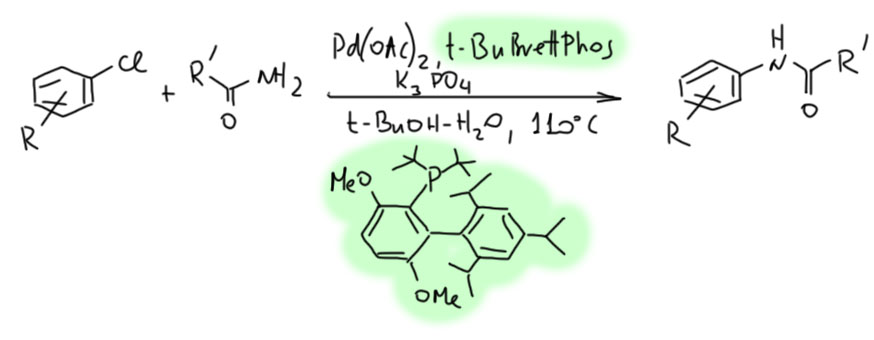
Восстановление Pd(2+) до Pd(0)
Палладий очень легко и очень многими способами восстанавливается до Pd(0). В отличие от никеля, кстати, поэтому в никелевом катализе предпочитают или брать Ni(0) сразу (например, Ni(COD)2) или добавляют востановители, например, или активированный цинк, или в последнее время стал очень моден металлический марганец, который, в отличие от цинка сам никуда не внедрится и ничего лишнего не восстановит.
А вот для палладия в большинстве случаев ничего брать не надо, поэтому в предкатализатор очень часто берут ацетат палладия. Хлорид берут очень редко, потому что он плохо растворим и лежит на дне, но его можно сделать растворимым очень просто, сделав комплекс с ацетонитрилом или бензонитрилом PdCl2(RCN)2. Лёгкость восстановления делает Pd(2+) часто лучшим источником Pd(0) чем готовые комплексы, особенно комплекс с dba. Многие исследователи и не раз замечали, что dba инлгда мешает, блокируя координационные места у палладия в тех случаях когда другие желающие их занять не проявляют достаточной настойчивости. Это не делает dba-комплексы плохими источниками палладия, но объясняет, почему их не используют просто всегда.
В кросс-сочетании восстановление происходит за счет окислительного гомо-сочетания нуклеофила (магний-, цинк-, олово-, борорганики – всё совершенно одинаково. Два переметаллирования и восстановительное элиминирование. Это очень лёгкая и быстрая реакция, а небольшая примесь гомо-продукта никому не мешает. 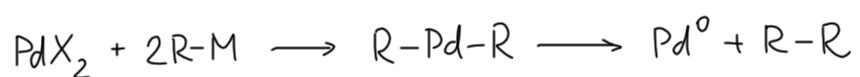
Если в реакционной смеси есть олефины, например, в реакции Мидзороки-Хека сам олефин и восстанавливает палладий. Можно нарисовать не один путь такого восстановления, но чаще всего предполагают обычный Вакер. Для этого в системе должно быть еще основание, но оно часто и так есть.
Еще один просто почти совсе универсальный путь – это бета-гидридное элиминирование. Оно работает и в Вакере, и в любой реакции, где может получиться комплекс палладия с лигандом, из которого идет такое элиминирование. Это алкилы, алкокси-группы, и многое из того, что можно получить из аминов. Поэтому, например, палладий восстанавливается любыми алкиламинами, в частности, триэтиламином. Они же и основанием послужат.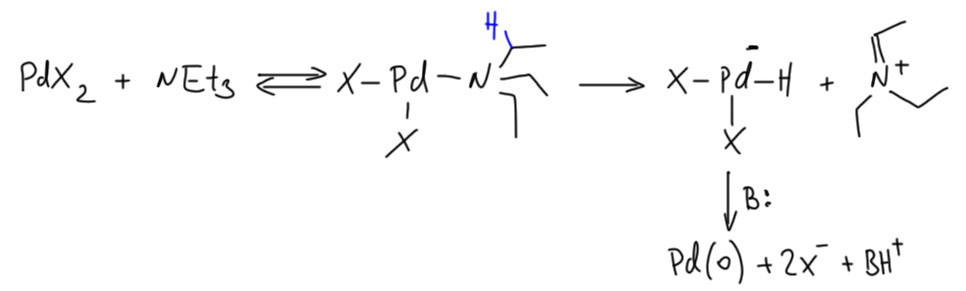
В реакциях с оксидом углерода восстановление происходит за счёт реакции водяного газа (water gas shift reaction, WGSR) или ее разновидностей – атаки нуклеофила на карбонильный лиганд. Проще всего она пишется с гидроксид-ионом, но и другие нуклеофилы годятся. Строго говоря, это еще один случай, когда работает бета-гидридное элиминирование.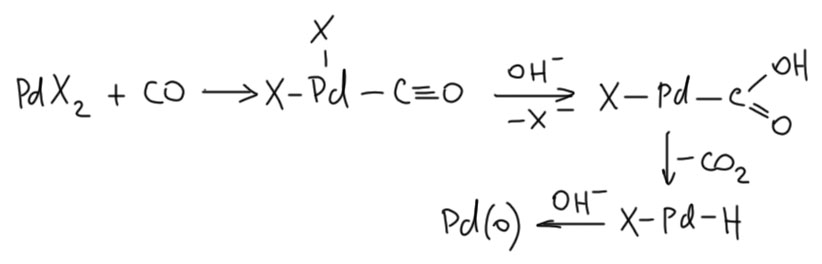
В реакциях диенов восстановление идёт через образование аллильных комплексов и их расщепление нуклеофилом. Мы не рассматривали таких реакций, потому что у них пока ещё слишком много проблем с селективностью.
Как видим, почти в любой реакции можно найти что-нибудь, что может востановить Pd(2+). По этой причине в палладиевом катализе не очень строго соблюдают правило хорошего тона в органическом синтезе – работай в инертной атмосфере даже если что-нибудь окисляешь, меньше будет побочных реакций, больше выход и селективность. Но в палладий-катализируемых реакциях, прежде всего в классическом кросс-сочетании, Хеке и карбонилировании воздух и правда почти не мешает.
Если ничего из перечисленного нет, а это бывает, например, в реакции Бухвальда-Хартвига с неалифатическими аминами, восстановителем может быть фосфиновый лиганд. Происходит что-то типа такого: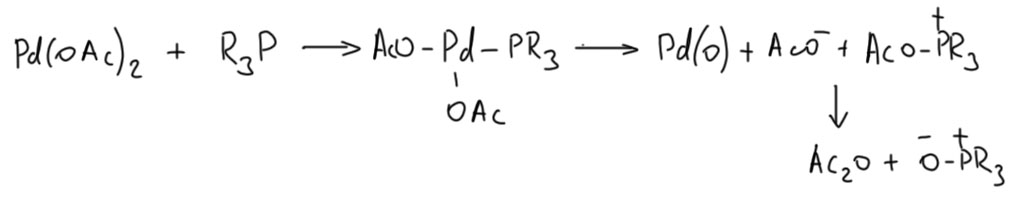 Уксусный ангидрид в условиях реакции аминирования немедленно превратися в ацетамид, и никто этого не заметит. Надо сказать, что это не очень быстрая реакция. В самом начале исследований аминирования особенно в системе Бухвальда – ацетат палладия с БИНАПом предактивация, по-видимому, осуществлялась именно так. И это не всегда было гладко, поэтому в методиках тех лет вы нередко увидите добавление фенилборной кислоты, которая собственно и восстанавливает палладий быстрее, чем фосфином. В наше время в аминировании предпочитают современные предкатализаторы аллильного типа или палладациклы Бухвальда G1,G2, G3, чтобы предактивация не была проблемой. В наше время вообще предкатализаторам стали уделять гораздо больше внимания, и это даёт более активные каталитические системы.
Уксусный ангидрид в условиях реакции аминирования немедленно превратися в ацетамид, и никто этого не заметит. Надо сказать, что это не очень быстрая реакция. В самом начале исследований аминирования особенно в системе Бухвальда – ацетат палладия с БИНАПом предактивация, по-видимому, осуществлялась именно так. И это не всегда было гладко, поэтому в методиках тех лет вы нередко увидите добавление фенилборной кислоты, которая собственно и восстанавливает палладий быстрее, чем фосфином. В наше время в аминировании предпочитают современные предкатализаторы аллильного типа или палладациклы Бухвальда G1,G2, G3, чтобы предактивация не была проблемой. В наше время вообще предкатализаторам стали уделять гораздо больше внимания, и это даёт более активные каталитические системы.
Что означают обозначения G1, G2, G3 над стрелками реакций?
Спасибо за вопрос. Это очень важная вещь, и она была в лекции, но как-то так получилось, что слайд потерялся. Я его восстановил в последней лекции в третьем блоке, где про CH-активацию и палладациклы. Это очень важный и один из двух чрезвычайно популярных типов предкатализаторов. Посмотрите слайд и пояснения под кнопкой Подробнее.
Как работает сверхкритический CO2?
Это очень правильный вопрос, потому что слайд про сверхкритический CO2 в его нынешнем виде – такое недоразумение, плод спешки и неуместного упрощения. Так хотелось показать немного парадоксальное родство этой среды с перфторорганикой, хотя это очень поверхностное сходство. В реальности, сверхкритическая углекислота действительно позволяет строить рециклизуемые каталитические системы, в том числе и фазо-разделяемые, но немного сложнее, чем водные или перфторированные.
Сверхкритический CO2 (обычно сокращают в scCO2) – среда, с одной стороны, довольно необычная, но с другой, вполне банальная. Жидкость это или газ? Жидкость, конечно, практически со всеми свойствами жидкости. Даже плотность у этой жидкости совсем обычная для жидкостей – в диапазоне 0.5 – 0.8 г/мл в зависимости от давления, на верхней границе жиапазона как у обычных углеводородов. А те свойства этой среды, которые ее отличают от жидкости, важны для физиков и физических химиков, но не для органиков.
В английском языке для обозначения сверхкритических сред используют очень удачную игру слов – в этом языке для обозначения жидкостей всегда было два слова – liquid и fluid. Первое как-то прижилось для обозначения обычных жидкостей в научно-техническом смысле, а второе болталось в области беллетристики и в научный контекст не проникало. А когда возникли сверхкритические среды, очень пригодилось, потому что как-то осталось ненагруженным. В русском языке проблема есть, потому что слово у нас одно – жидкость, второго в загашнике не оказалось, пришлось заимстововать немного корявое флюид, которое вполне себе существовало, но в слое архаично-возвышенной лексики с сильным ароматом выпендрёжа. Этот аромат за ним и тянется. Зато короткое.
Для органиков это именно жидкость. Важнейшее свойство жидкости для органиков – растворять другие вещества и обеспечивать быструю диффузию их молекул, не ограничивающую вероятности столкновения и реакции – в этой среде есть в полной мере. Поскольку это среда еще и с очень низкой вязкостью, диффузия в ней совсем быстра и это хорошо. Особенно это хорошо для использования в проточных реакторах, где низкая вязкость позволяет легче и быстрее двигаться через всякие затейливые конструкции с высоким гидравлическим сопротивлением.
Есть ли в этой среде сольватация – а как же, конечно есть, но это неполярный растворитель (в этом смысле немного похожий на четырёххлористый углерод), но имеющий достаточно поляризованные связи и атомы кислорода с небольшой основностью Льюиса (в этом смысле это немного похоже на ацетонитрил). Поэтому это слабосольватирующий растворитель. Это иногда хорошо (мы знаем, что сольватация иногда мешает реакциям, потому что реагентам нужно как-то избавиться от молекул растворителя чтобы прореагировать). А иногда плохо, потому что в этой жидкости далеко не все растворяется, и с этим есть много проблем. Но к ней можно добавить другие органические растворители, и в таких смесях (например scCO2 – метанол) растворяются и довольно полярные соединения.
Крайне необычным свойством сверхкритических сред и scCO2 в том числе – отсутствие газовой фазы. С такими средами по понятным причинам работают в замкнутых сосудах под давлением. И есть такая вещь как жидкий CO2 – и если у нас именно он, мы можем заглянуть в сосуд (окошки иногда делают в сосудах для работы с такими средами) и увидеть жидкость, мениск, и над мениском газовую фазу. Но как только мы дойдем до критической точки (а у углекислоты она очень близко – температура около 30 градусов и давление около 70 атмосфер), мы увидим, что мениск исчез, а сосуд наполнен как будто прозрачным газом (наши мозги отказываются понимать отсутствие мениска по другому – грубо говоря, если ничего не плещется, то это не жидкость). Но это именно скорее жидкость, а газовой фазы нет потому что в этом и есть сущность этого явления – исчезновение двух отдельных фаз и появление одной, новой.
У этого свойства есть одно очень практическое следствие – если нет газовой фазы, то любые газы идеально смешиваются с сверхкритической средой. Это совершенно очевидное свойство – ведь если бы мы нашли газ, которые не смешивается с сверхкритической средой, возникла бы поверхность раздела фаз, и в образовавшейся газовой фазе – парциальное давление CO2 – газообразного. А это противоречит самой сути сверхкритической фазы. Это свойство – идеальная смешиваемость с газами – очень любят в реакциях с участием газов – гидрировании, гидроформилировании, и т.п. Эти реакции очень много исследовали в scCO2.
А вот наличие настоящей жидкой фазы в сосуде с scCO2 вполне возможно, потому что многие жидкости с ней не смешиваются. И это ничему не противоречит, потому что такая жидкая фаза содержит малую концентрацию растворенной углекислоты и никак не вмешивается в фазовую диаграмму самой углекислоты. Поэтому с сверхкритической углекислотой вполне возможно составление двухфазных систем, играющих на распределении молекул растворенных веществ между фазами. В реальности таких систем очень мало, потому что играть на таких переносах пока не очень получается. Поэтому и картинка в слайде, сделанная по аналогии с водными и перфторорганическими системами, и правда скорее мечта, чем реальность.
У сверхкритических фаз очень высокая летучесть. Собственно, это совершенно очевидно, ведь отсутствие газовой фазы делает такие среды свободно распространяющимися за пределы сосуда, как только такая возможность в виде открытой заслонки им будет предоставлена. Только нужно так проектировать прибор, чтобы при выходе среды за пределы исходного сосуда давление не упало ниже критического, но это решаемая инженерная задача. И у такой жидкости есть очень интересное свойство захватывать с собой растворенное вещество. Собственно, оно так и должно быть, – куда, собственно оно должно из нее деться – но выглядит со стороны это так, как будто такая жидкость летит и делает летучими другие вещества, даже такие, которые просто так в этих условиях никогда не перегонялись бы. Это очевидное, но совершенно невероятное явление было случайно обнаружено в лаборатории Карла Циглера вскоре после войны, когда там открыли то, что позже назвали полимеризацией Циглера-Натты и оценили Нобелевской премией. У них там был сверхкритический этилен, и они обнаружили, что в приборе, где изучали полимеризацию, маслянистые капли олигомеров этилена оказывались очень далеко от реактора, хотя при той температуре, которая была в реакторе, эти вещества никак не могли туда долететь. Из этого наблюдения через пару десятков лет родился процесс, запатентованный в институте, которым руководил Циглер – продажа лицензий на этот процесс принесла институту Исследования Угля совершенно колоссальные деньги. Этот процесс – извлечение кофеина из сырых бобов кофе с помощью сверхкритической углекислоты в производстве декаффеинированного кофе и собственно кофеина. В этом процессе бобы буквально замачивают в scCO2 и через некоторое время раствор кофеина в scCO2 подают на непрерывный экстрактор, где кофеин из сверхкритической углекислоты эктрагируют горячей водой, в которой кофеин растворим гораздо лучше чем в неполярной и неспособной давать водородные связи углекислоты. Из экстрактора scCO2 возвращают к бобам – растворимость кофеина невелика и процесс должен быть непрерывным, чтобы получить высокую степень извлечения. Конечно, при этом частично в воду переходит и углекислота, но не очень много, так как хотя там и довольно высокое давление (увеличивает растворимость), но и довольно высокая температура (уменьшает растворимость). После экстракции водный раствор кофеина отправляют на концентрирование и кристаллизацию, а выделяющуюся газообразную углекислоту сжимают и возвращают в экстракцию, чтобы не было потерь. Основная часть углекислоты в этом процессе постоянно остается в состоянии сверхкритической жидкости, что делает процесс экономичным и безопасным, а кофе-бобы после такой экстракции не приобретают никаких посторонних вкусов и запахов (немцы сделали даже такой эксперимент – взяли радиоактивную углекислоту и убедились, что после окончания экстракции кофе не содержит даже следов радиоактивности), и после обжаривания дает нормальный кофейный аромат. По сравнению с другими способами экстракции кофеина (водой, органическими растворителями) этот метод практически не влияет на качество кофе.
Этот реальный промышленный процесс показывает две важные особенности scCO2 – способность захватывать растворенные органические вещества и возможность самой обычной двухфазной экстракции без нарушения сверхкритичности этой среды. Поэтому, ничего невозможного в реализации рециклизации нет и в катализе – нужно только придумать, как развести по разным фазам катализатор и продукт. Пока что в большинстве реализаций предпочитают упрощать задачу, используя иммобилизованные катализаторы на подложках – тогда катализатор остается в реакторе, а продукты реакции вместе с scCO2 уводят в аппарат, где углекислоту и продукты разделяют тем или иным способом, например, той же экстракцией растворителями, не смешивающимися с сверхкритической жидкостью.
Пока на этом остановимся. Со временем я сдлеаю более серьезный апдейт соотвествующего слайда и сделаю к нему пояснения с примерами.
Норборненовые каскады
Каскады с участие норборнена (каскады Кателлани) действительно производят ошеломляющее впечатление головокружительным мельтешением интермедиатов. Кажется, что так не может быть, и что в каждой стадии возможны другие пути. Но такое впечатление производят любые сложные каскады. Каскады интересны тем, что заранее предсказать, что получится, никогда нельзя. И большинство кандидатов в каскады завершается невообразимой кашей продуктов. Исследователь находит хороший прототип, с которым происходит нечто необычное, и дальше просто исследует – месит реакционные смеси, разбирается в продуктах. И если исследователь упорен и удачлив, иногда получаются шедевры, а каскад Кателлани с завершением кросс-сочетанием – несомненно шедевр.
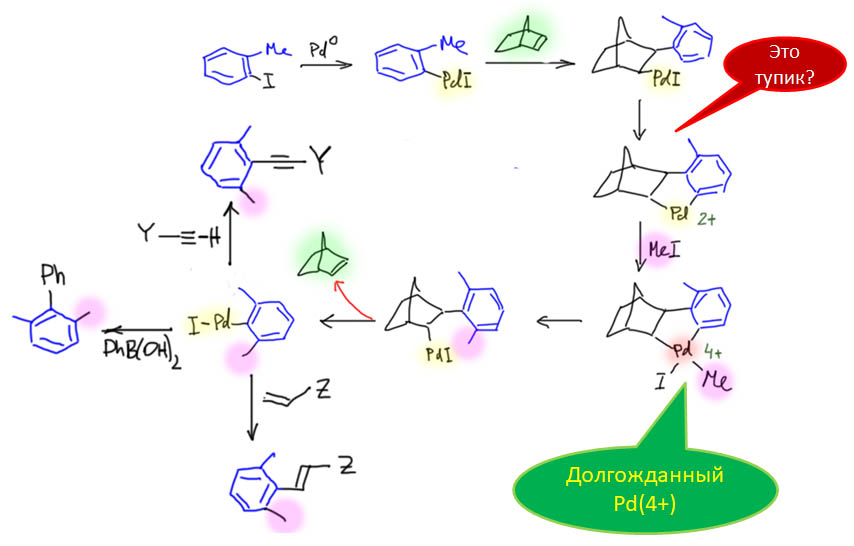
В данном случае реакционная смесь сразу содержит все компоненты, включая бороновую кислоту и основание, необходимые для завершающего кросс-сочетания (Catellani, M.; Motti, E.; Minari, M. Symmetrical and
Unsymmetrical 2,6-Dialkyl-1,1′-Biaryls by Combined Catalysis of Aromatic Alkylation via Palladacycles and Suzuki-Type Coupling. Chem. Commun. 2000, 157–158). Но в реакции получаются почти исключительно каскадные продукты, а побочные тоже получаются преждевременным завершением каскада, но не простой реакцей Судзуки-Мияуры из исходного иодпроизводного и бороновой кислоты.
Это означает только одно – скорость карбопалладирования выше скорости переметаллирования. Аддукт окислительного присоединения сразу подхватывает весьма донорный олефин, и это не даёт случиться переметаллированию. А как только карбопалладирование случилось, дальше это колесо должно и будет крутиться пока не упрётся в обратную экструзию норборнена, и тут уже получается комплекс, который по стерическим причинам больше не входит к карбопалладирование (он только что из него вышел) и медленно идёт в кросс. Справедливости ради заметим, что реакцию ведут при комнатной температуре и очень медленно, а если бы вы захотели сразу кросс-сочетание, то просто повысили бы температуру – в этих условиях связывание олефина сразу стало бы обратимым и очень вероятно, что вместо каскада получилось бы кросс-сочетание с осложнениями.
Бета-Si элиминирование вместо гидридного - почему?
Имеется в виду вот этот каскад от одного из главных каскадёров мировой химии Луца Тице (Lutz Tietze et al. Angew. Chem. 2013, 125, 3273 –3276). Каскад завершается не вполне обычным образом – вместо гидрида палладия отскакивает, как может показаться, кремниевый комплекс палладия. Впрочем, обычные продукт завершения Хека тоже образуется, но в небольшом количестве (13% против 76% основного продукта). Что здесь произошло и как это соотносится с широко разрекламированной нездоровой тягой палладия именно к гидриду? Разве кремний может заменить гидрид? А известна вообще такая реакция?
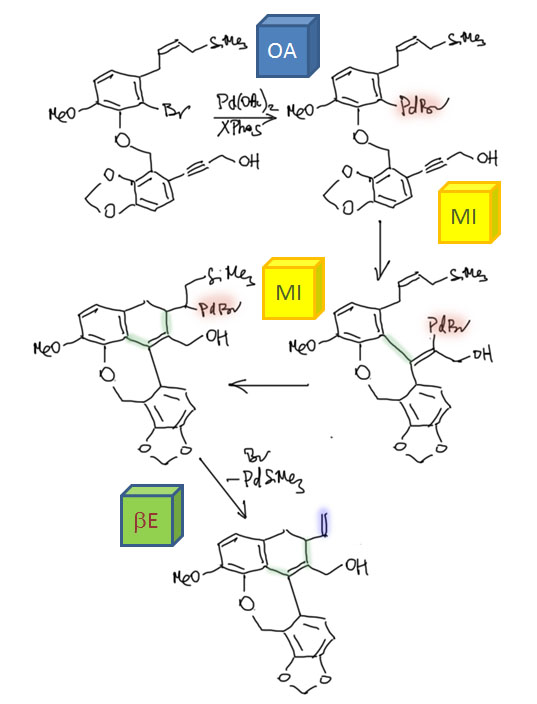
Известна, и даже не одна такая реакция. Это занятная история, которая ещё раз показывает, как красивые идеи в химии переходных металлов изменяются до неузнаваемости по мере встречи их, идей, с экспериментом. А потом и вовсе куда-то деваются. Впрочем, почти всегда что-то интересное получается, если автор идеи не упирается рогом, и не требует, чтобы всё было как задумано, или пусть провалится, – а следует за экспериментом, изобретая новые интерпретации и приспосабливая реальные результаты к решению иных задач.
Это исследование восходит к давней идее Тице, как победить, хотя бы иногда, назойливое блуждание двойной связи в реакции Хека, когда элиминирование гидрида может происходить с двух сторон. Это блуждание убивает и региоселективность и стереоселективность, в тех случаях, когда она может возникнуть. А в лаборатории Тице очень активно разрабатывали методологию внутримолекулярного Хека, как средства быстрого построения очень сложных структур. И обратимое гидридное элиминирование очень часто портило первоначальный блестящий замысел. Тице возненавидел эту реакцию. Он пробовал разные условия в Хеке, разные модные, как тогда считалось, катализаторы, в том числе палладацикл Херманна-Беллера, и разные другие примочки, да всё без толку. И однажды он решил, что если вводить в Хека аллилсиланы, то кремний будет помогать β-гидридному элиминированию. В этом случае оно будет происходить быстро, и дальше можно будет так подобрать условия, чтобы гидрид палладия быстро превращался в Pd(0) и не отсвечивал в равновесных присоединениях-отщеплениях. 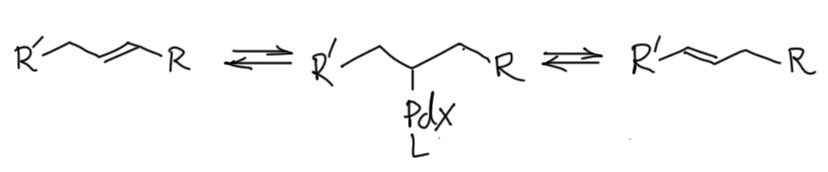 Почему Тице так решил? Это очень непросто понять, потому что он сам руководствовался идеей, что кремний ускоряет гидридное элиминирование именно со своей стороны. Представим, что один из заместителей – кремниевая группа. Тогда мы можем представить, что подвижность атома водорода под кремнием увеличивается. Почему? Ну, во-первых, кремниевые группы считаются объёмистыми, а значит, по стерическим причинам с этой стороны водород будет большн подставляться под син-взаимодействие с палладием. Во-вторых, нам может прийти в голову, что кремний увеличивает кислотность этого атома, по аналогии с тем, как это делает сера (в хорошо всем известном примере повышенной кислотности 1,3-дитианов – элементы 3 периода вообще известны этим эффектом, который обычно объясняют σ*-π сопряжением). В этом месте вас может поразить вполне очевидная идея, что это странное объяснение – ведь палладий охотится на гидрид, а вовсе не на протон, и ему был бы более угоден повышенный гидридный статус атома водорода, чем кислотность. А c гидридным статусом там как раз всё плохо – способность к отщеплению гидрида всегда коррелирует со стабилизацией образующегося на этом месте карбокатиона, но кремний как раз карбокатион под собой дестабилизирует.
Почему Тице так решил? Это очень непросто понять, потому что он сам руководствовался идеей, что кремний ускоряет гидридное элиминирование именно со своей стороны. Представим, что один из заместителей – кремниевая группа. Тогда мы можем представить, что подвижность атома водорода под кремнием увеличивается. Почему? Ну, во-первых, кремниевые группы считаются объёмистыми, а значит, по стерическим причинам с этой стороны водород будет большн подставляться под син-взаимодействие с палладием. Во-вторых, нам может прийти в голову, что кремний увеличивает кислотность этого атома, по аналогии с тем, как это делает сера (в хорошо всем известном примере повышенной кислотности 1,3-дитианов – элементы 3 периода вообще известны этим эффектом, который обычно объясняют σ*-π сопряжением). В этом месте вас может поразить вполне очевидная идея, что это странное объяснение – ведь палладий охотится на гидрид, а вовсе не на протон, и ему был бы более угоден повышенный гидридный статус атома водорода, чем кислотность. А c гидридным статусом там как раз всё плохо – способность к отщеплению гидрида всегда коррелирует со стабилизацией образующегося на этом месте карбокатиона, но кремний как раз карбокатион под собой дестабилизирует. 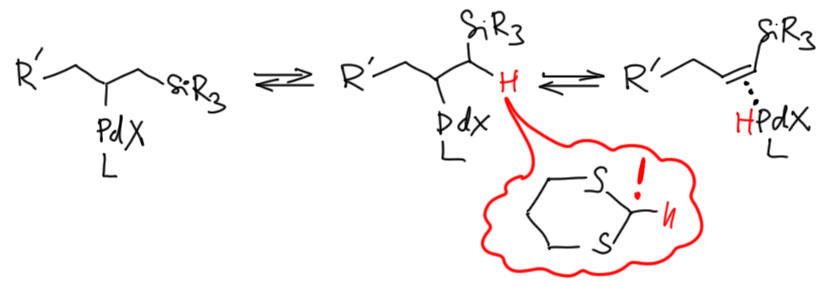
Не беда. Химия хороша тем, что при известной ловкости рук, многие её законы напоминают дышло гораздо даже больше, чем законы человеческие. Этим искусством – приспособления законов химии под нужды конкретных задач – нужно владеть, но при этом надо обязательно держать себя в руках, чтобы не плодить совсем уж сказок – объяснение должно оставаться правдоподобным и по возможности согласовываться с экспериментальными данными. Химия переходных металлов этому очень способствует, ведь в ней царят согласованные механизмы, основной чертой которых является адаптивность, приспособляемость к реальности. Палладий забирает гидрид, но ведь это формально, просто потому что принято считать этот лиганд гидридом, а как там перераспределяются электроны в переходном состоянии, когда водород отходит к палладию, который переползает с σ-связи на дигапто-связь (как мы знаем, она тоже имеет σ-характер, но это не важно). Поэтому можно ожидать, что кремниевый заместитель действительно облегчает β-гидридное элиминирование. Но когда дело дошло до экспериментального воплощения этой идеи, всё получилось ещё интереснее – в зависимости от состава каталитической системы получалось или именно то, что хотел Тице, или нечто иное – уходил вместо гидрида кремниевый остаток (L. F. Tietze, R. Schimpf, Angew. Chem., Int., Ed. Engl., 1994, 33, 1089). 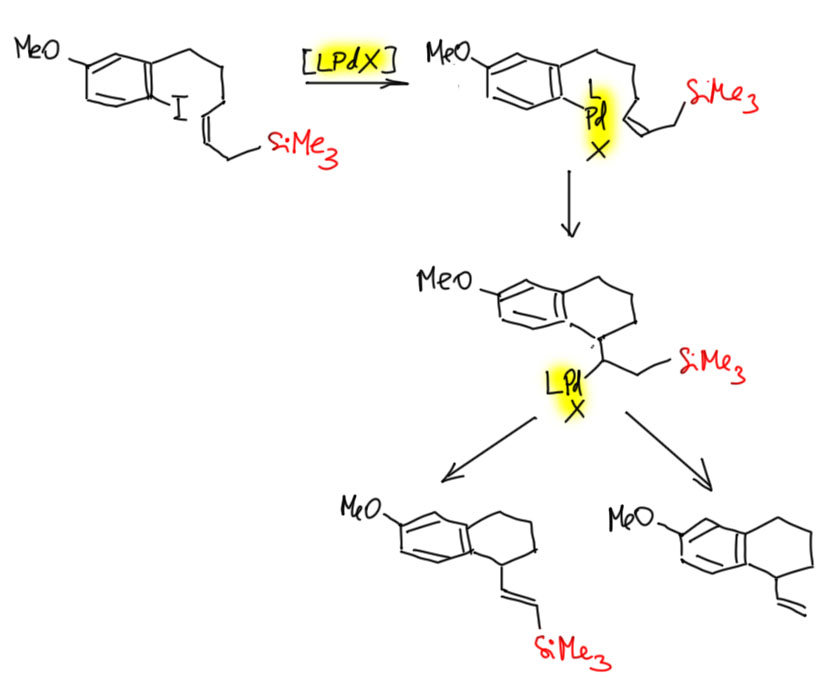
Направление реакции зависит от каталитической системы. Обычная для Хека система неселективна – даёт и то, и то, и еще кучу побочных, зато быстро. Селективность возникает, во-первых, тогда когда используется не просто основание, но основание, содержащее ионы серебра. Сейчас разберёмся, что они делают. Обычный фосфин даёт практически количественно то, что и хотел Тице – селективное элиминирование из-под кремния без миграции двойной связи. А вот использование дифосфина привело к элиминированию кремниевого остатка, к сожалению, не количественному, но в количестве, которое обычно характеризуют как высокую селективность. В придачу, поскольку лиганд был взят хиральный, произошёл и весьма примечательный перенос хиральности – внутримолекулярное карбопалладирвоание произошло энантиоселективно. 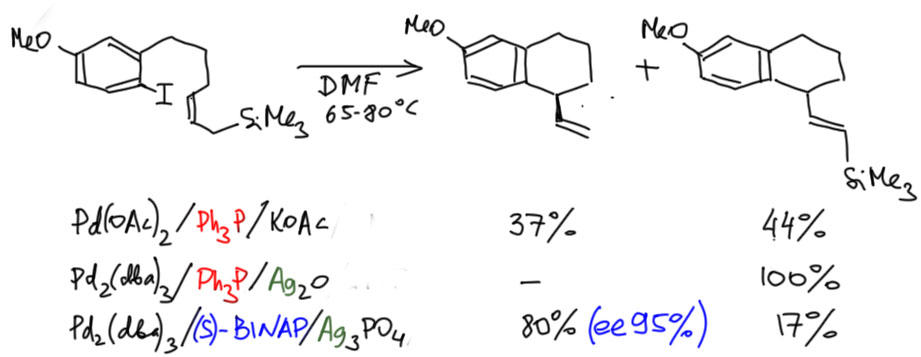
Вопрос первый – что делают соли серебра в Хеке? Надо было бы раньше это обсудить, поскольку это очень распространенный приём, но хоть здесь. Соли серебра удаляют галогенид из координационной сферы палладия, оставляя пустое место (ясно, что занятое сильно-лабильным неявным лигандом, скорее всего, растворителем). Возникает, во-первых, положительный заряд на палладии. Считается, то это приводит к значительному усилению льюисовой кислотности (акцепторности, электрофильности) переходного металла. И кроме того – и это очень важно – освобождают координационное место для следующих стадий каталитического цикла. И таким, образом, в частности, позволяют использовать в Хеке бидентатный фосфиновый лиганд (напоминаю, что пока на палладии висит галогенид, а это, особенно иодид, лиганд очень прочно связанный, мы не можем делать Хека – не хватает места в координационной сфере).
Теперь разберёмся в тем, почему в такой каталитической системе произошло то, что хотел Тице – селективный уход водорода из-под кремния. Правильного ответа на это не знает никто, но нужно выработать гипотезу, которая как-то учитывает увеличение кислотности протона. Одна из возможностей – реанимировать старый спор, не может ли β-гидридное элиминирование хотя бы иногда происходить не как согласованный процесс син-элиминирования, а как обычный E2-процесс с участием основания. Если это возможно, то тогда понятно, почему в этом случае нам понадобилось достаточно сильное основание (оксид серебра, как правило, используют в так называемой “влажной” форме, фактически в виде гидроксида серебра, а это в ДМФА будет очень неслабым основанием).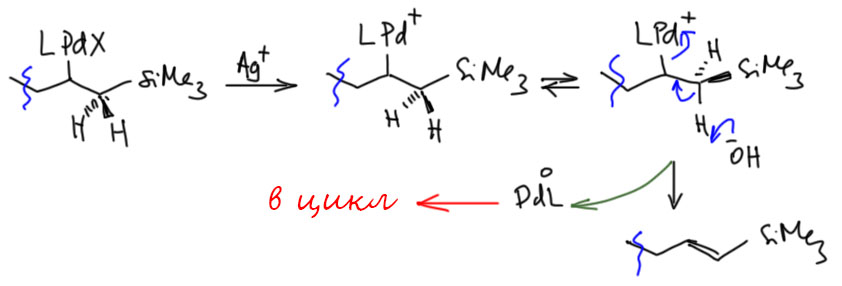
Но если такое элиминирование идёт, как же быть с требованием син-элиминирования, на котором в основном строится вся каскадная химия? Да, нехорошо получается. Но мы всегда можем сказать так – это исключение, которое происходит только тогда, когда по каким-то причинам не идёт (или идёт медленно) обычное β-гидридное элиминирование, или атом водорода имеет повышенную кислотность. Впрочем, дальше нас ждёт еще одно испытание, когда мы окончательно запутаемся. А почему в системе с дифосфином уходит кремний? Тице объясняет это тоже E2-элиминированием, такой разновидностью, когда вместо протона уходит кремниевый остаток. С точки зрения свойств уходящих групп, кремний такой же электрофуг как протон – уходит, оставив пару электронов на месте. И если протон подхватывает основание, кремний подхватывает любой жёсткий нуклеофил с основным кислородом. Вон там у Тице это фосфат (я не буду выписывать фосфат, напишу в общем виде OZ). И обратите внимание, я сразу написал плюс на палладии, потому что ион серебра и здесь очистил координационной место.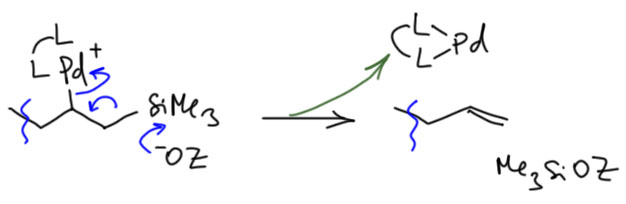
Так, чёрт возьми, а почему эти две системы так сильно отличаются? Ну, с дифосфином так и хочется сказать-написать, что мы же знаем, что дифосфин блокирует отщепление гидрида. Но сразу возникнет ощущение, что мы заврались – ведь серебро очистило нам местечко. И да, и в случает с трифенилфосфином мы же отказались от β-гидридного син-элиминирования. Заврались-заврались, и как выходить из этого неловкого положения непонятно. А что там у Тице? У Тице всё совсем грустно – я хотя бы немного подчистил его неряшливые объяснения, но, как видите – тоже не очень преуспел. Такова она, наука химия, – даже если что-то работает, не всегда поймешь почему.
Давайте посмотрим, как эта система работает в других работах. Во-первых, для того чтобы на палладии было свободное место и положительный заряд, можно вместо удаления галогенида солью серебра сразу взять трифлат (L. F. Tietze, A. Modi, Eur. J. Org. Chem. 2000, 1959-1964). В этом случае вполне работает обычная каталитическая система с трифенилфосфином – кремний сохраняется. Можно сделать вывод, что с таким палладием вполне работает гипотеза об облегчении β-гидридного элиминирования. Остается непонятным, идёт ли это элиминирование как син-элиминирование или как E2, но использование слабого основания, обычного для классического Хека скорее склоняет к выводу, что это именно син-элиминирование, но по какой-то причине палладий с положительным зарядом предпочитает выцеплять более кислый водород из-под кремния.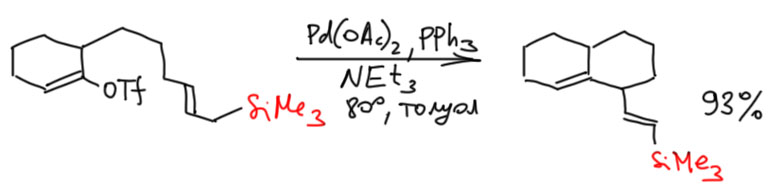
Дальше, даже этот рубеж будет успешно оставлен. Тице начал использовать реакцию в каскадах.И уже довольно простой каскад, состоящий из двух карбопалладирований, идёт в самой обычной стандартной системе (зачем в Хеке используют соли четвертичного аммония – так называемый протокол Ларока-Джеффери – обсудим как-нибудь отдельно, но это точно не имеет никакого отношения к проблеме возможного элиминирования водорода или кремниевого остатка) без удаления галогенида, и приводит к элиминированию гидрида и сохранению кремния (L. F. Tietze, K. Kahle, T. Raschke, Chem. Eur. J. 2002, 8, 401-407).
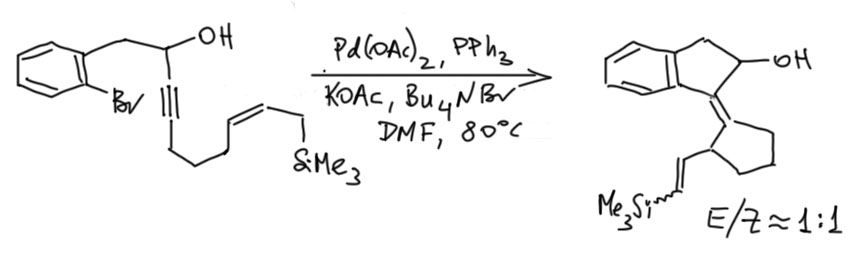
Цикл каскада включает карбопалладирование тройной связи, затем двойной, которое может осуществиться только в такой конфинурации как показано, поэтому геометрия соединения двух колец на двойной связи очень точно задана – это диастереоспецифическая реакция в этом месте. Увы, β-гидридное элиминирование не может похвастаться не то что диастереоспецифичностью, но и хоть какой мизерной диастереоселективностью, но по отношению к альтернативе – отщеплять водород или силильный остаток – работает очень хорошо, побочный продукт без кремния образуется в очень малых количествах.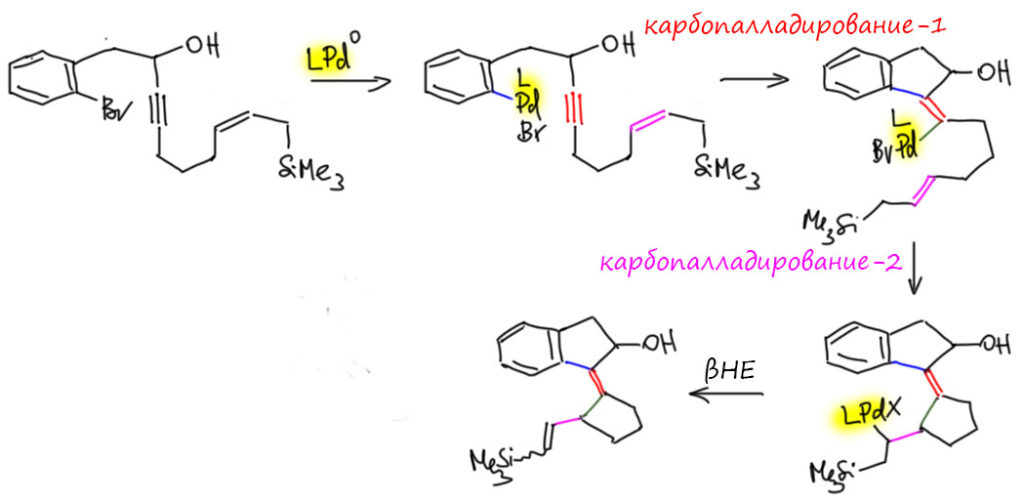
И вот мы подходим к каскаду, который вызвал вопрос. И вновь всё идёт не так, как было задумано. Для каскада пробовали несколько каталитических систем, но 2013 уже было грех не попробовать модные лиганды Бухвальда, и действительно, XPhos, очень объёмистый и донорный лиганд, сработал лучше всех. Обратите на это внимание – каскад производит впечатление реакции, в которой палладиевый комплекс путешествует по большой структуре, как кузнечик по листу подсолнуха – кажется, что палладий вместе со своими лигандами должны быть маленькими и юркими, иначе где-нибудь застрянет, не пролезет. А теперь представьте себе палладий с XPhos – да эта штука едва ли не больше той молекулы, по которой ей выпало ползать, сшивая новыми связями кольца и фрагменты! Я даже на схеме был вынужден сократить все вокруг палладия, чтобы не захламлять, и просто показываю жёлтым, что палладиевый центр везде один и тот же – с огромным лигандом и бромидом. Это, видимо, должно, скорее напоминать такую большую машину, в которую вползает исходная молекула, там что-то шумит, булькает, трещит, грохочет – вот уже выползает с другой стороны готовая. Но кроме размера, мы не можем не обратить внимание на другое свойство – сильнодонорный лиганд делает палладий скорее высоконуклеофильным, хотя до сих пор у нас все время было что-то повышенно кислотное, электрофильное. И уходит у нас в этом случае не гидрид, а именно кремний, очевидно, путём обычного элиминирования, с содействием ацетат-иона (реакция идёт в присутствии ацетата тетрабутиламмония, то есть в ситуации максимального увеличения нуклеофильности ацетат-иона). Да, можно сказать, что гидрид и не мог уйти – XPhos и бромид блокируют координационные места, необходимые для захвата гидрида. Разумно, хотя такой продукт всё же получается в количестве 13%. 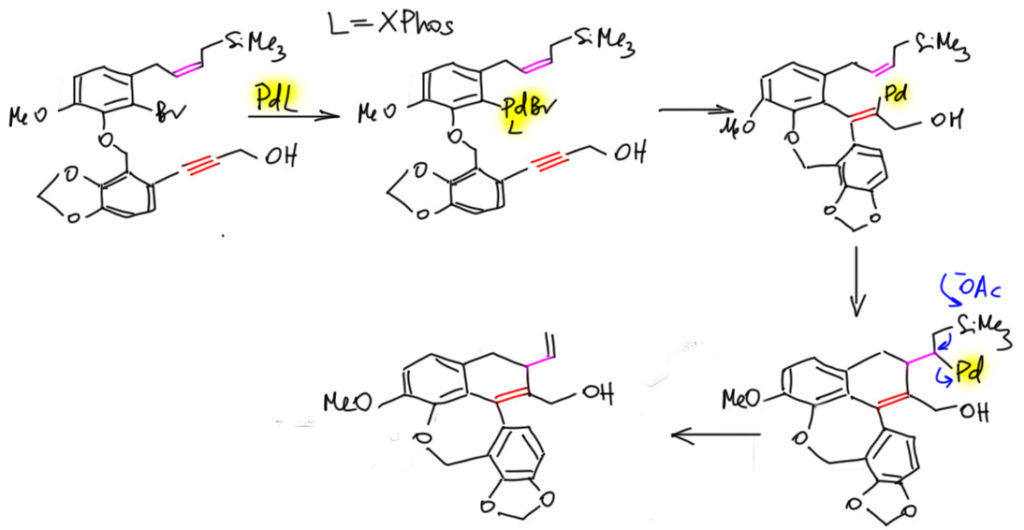
Таких примеров в литературе можно найти ещё с полдюжины. Можно сделать вывод, что уходить может как гидрид, так и силильный остаток, и закономерности очень запутанны, настолько что предсказать, что в какой ситуации уходит, можно только заранее зная результат.
Силил-Хек
А теперь посмотрим на проблему с другой стороны. А может ли вообще образоваться силильный комплекс палладия в результате элиминирования, похожего на классическое β-гидридное элиминирование? А как это узнать? А, как водится в химии переходных металлов, посмотреть, не существует ли обратной реакции, миграционного внедрения олефина по связи Pd-Si – это будет называться, видимо, силапалладированием. Оказывается, вполне себе существует, и есть очень интересный аналог реакции Хека, в которой вместо связи C-галоген используется связь кремний-галоген. Такую реакцию впервые описал Дональд Уотсон с сотрудниками совсем недавно (J. R. McAtee, S. E. S. Martin, D. T. Ahneman, K. A. Johnson, D. A. Watson Angew. Chem. Int. Ed. 2012, 51, 3663 –3667). Почему недавно? А потому что реакция весьма хитрая, и для неё нужно было уже здорово разбираться в том, как можно способствовать необычным реакциям окислительного присоединения, миграционного внедрения и элиминирвоания, и иметь большой выбор современных лигандов и пред-катализаторов. С инструментами 20 века к такой задаче не подступиться. Итак, задача – построить кремниевый аналог реакции Хека. В конце, вслед за Уотсоном, запишем два возможных продукта β-гидридного элиминирования, сделав вид, что мы ничего не знаем про исследования Тице, которые вроде бы однозначно говорят, что водород должен уходить из-под кремния. И, возможно, нас ждёт ещё один сюрприз.
Каталитическая система была найдена. Во-первых, потребовался высокоэффективный предкатализатор, и здесь пригодился очень интресный комплекс палладия с двумя алкильными лигандами. Обычно такие комплексы должны быть нестабильны из-за очевидной склонности с восстановительному элиминированию, но если взять два объёмистых алкила, кремниевых аналога неопентила, то комплекс устойчив при комнатной температуре, хотя при небольшом нагревании в присутствии объёмистого анциллярного лиганда сбрасывает алкан и даёт монолигандный комплекс Pd(0). 
Этот самый анциллярный лиганд пришлось поискать. Он должен и обеспечивать окислительное присоединение к связи Si-галоген, для начала хотя бы Si-I, и всё остальное, в том числе миграционное внедрение кремниевого комплекса (силапалладирование). Первый такой лиганд оказался необычно прост, – трет-бутилдифенилфосфин. Все остальные условия очень похожи на классического Мидзороки-Хека. Получилась такая реакция со стиролами. 
А вот если алкен подлиннее (не пóдлиннее, а подлиннéе) и возникает альтернатива, то, во-первых, потребовалось немного изменить протокол и взять очень похожий, но специально сделанный для этой цели лиганд JessyPhos (по моде, заведённой Бухвальдом, Уотсон назвал новый лиганд по имени сотрудника, который его сделал – Джесси Мак-Эйти)(Jesse R. McAtee, G. P. A. Yap, D. A. Watson J. Am. Chem. Soc. 2014, 136, 10166-10172). И получается при этом исключительно продукт гидридного элиминирования не из-под кремния, а с другой стороны (в некоторых случаях с очень малой примесью второго продукта). А как же идеи Тице? Да кто ж знает! В этом вся химия. Может быть лиганд виноват, что всё так изменилось. А может и не было никаких идей, а так, померещилось…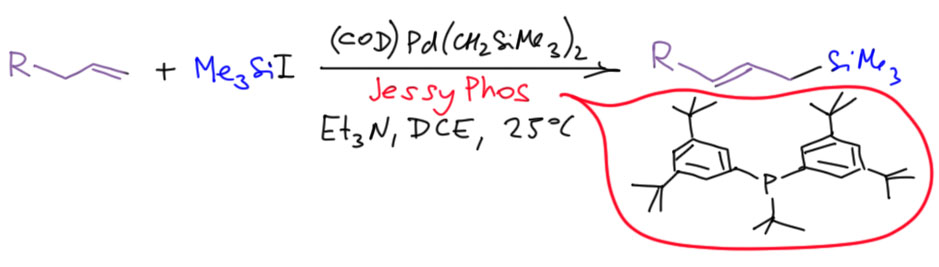
Ионные жидкости и NHC-карбены
Великолепный вопрос! Точно такой же возник и у меня, когда я впервые столкнулся с работами по использованию ионных жидкостей в катализе. Действительно – это совпадение или промысел уполномоченных богов, что самыми популярными ионными жидкостями являются соли имидазолия. Депротонирование солей имидазолия действительно даёт карбены. И что – участвуют они или нет? Вопрос кажется почти риторическим – а куда, типа, они денутся? Удивительно, но ответить на этот вопрос прямо невозможно, потому что это, кажется никто прямо не исследовал. Поэтому разделим вопрос на две части – практическую и демагогическую. Практическая часть – как работают ионные жидкости и как с ними можно устроить рециклизуемую каталитическую систему. В демагогической – попробуем вернуться к вопросу, могут ли они помогать катализу не как растворители, а как лиганды или как органокатализаторы.
И что – участвуют они или нет? Вопрос кажется почти риторическим – а куда, типа, они денутся? Удивительно, но ответить на этот вопрос прямо невозможно, потому что это, кажется никто прямо не исследовал. Поэтому разделим вопрос на две части – практическую и демагогическую. Практическая часть – как работают ионные жидкости и как с ними можно устроить рециклизуемую каталитическую систему. В демагогической – попробуем вернуться к вопросу, могут ли они помогать катализу не как растворители, а как лиганды или как органокатализаторы.
Ионные жидкости это просто расплавы солей. Можно расплавить хлористый натрий и получить ионную жидкость, только для органики она не очень полезна, так как органика не любит высоких температур, да и не будет в этом растворяться. Поэтому для органики подбирают соли с невысокими температурами плавления, причем такие, которые имеют в структуре углеводородные хвосты, что придает таким средам способность смешиваться с малополярной органикой. В начале это были банальные четвертичные соли аммония – если на азоте прямые алкильные хвосты, они создают конформационный беспорядок и мешают кристаллизации. Уже такая распространенная соль как бромид тетрабутиламмония плавится при 103ºС, что даёт жидкость в интервале температур, вполне пригодном для множеста органических реакций. Но еще лучше когда соль является жидкой уже при комнатной температуре. И таких тоже полно. Абсолютными чемпионами по популярности являются продукты кватернизации имидазола, а среди них N-бутил-N-метилимидазолий с разными противоионами, что сокращается как BMIM[противоион], и противоионы бывают как самые простые типа бромида или тетрафторбората, так и более сложные типа NTf2. Такие противоионы дают более широкие диапазоны жидкого состояния, и не мешают, так как не обладают собственной нуклеофильностью.
Пионерской работой по применению ионных жидкостей в катализе является статья британцев Томаса Уэлтона с сотрудниками (C. J. Mathews, P. J. Smith, T. Welton Chem. Commun., 2000, 1249), и в той статье в общем есть почти всё, о чём можно говорить в этом контексте. Во-первых, ионные жидкости повзоляют применять самые обычные методы работы без модификаций, в ионных жидкостях растворяются как органические реагенты, так и соли, хотя для твёрдой органики требуется более длительное перемешивание для растворения, а с солями иногда приходится добавлять их не в твёрдом виде, а в виде раствора в минимальном количестве воды. Дальше все обычно – перемешиваем и нагреваем. Так была сделана стандартная реакция Судзуки-Мияуры.
Реакция прошла – как выделять продукт. Для ионных жидкостей есть несколько путей. Во-первых, продукт можно экстрагировать органическим растворителем. Многие растворители смешиваются с ионными жидкостями, но очень хорошо известно, что четвертичные аммонийные соли, которыми и являются большинство ионных жидкостей, не растворяются в эфире и других рстворителях эфирного типа. Четвертичные аммонийные соли всегда высаживают из растворов эфиром. И эфир не смешивается с ионными жидкостями аммонийного типа. Эфир – хороший, хотя и не универсальный растворитель для экстракции, он отлично растворяет многие не очень большие органические соединения, и неплохо экстрагирует, в частности, продукты кросс-сочетания. Палладиевые комплексы не экстрагируются и остаются в ионной жидкости.
Второй способ использует пренебрежимо малую летучесть ионных жидкостей и их высокую устойчивость к нагреванию. Запас по температуре достигает 300ºС, причём возможно удаление при пониженном давлении. Палладиевые комплексы опять остаются в ионной жидкости.
Третий способ предполагает контролируемое разбавление водой при небольшом нагревании, при этом вся органика, содержащаяся в больших количествах (продукты реакции) выпадает, это особенно удобно если продукты кристаллические. После такой процедуры систему можно охладить, она опять расслоится на водную и ионно-жидкостную фазу (дальше буду сокращать в ИЖФ), продукт отфильтровать, и при необходимости остаточную воду удалить нагреванием в вакууме. Здесь нужно прояснить, как ионные жидкости относятся к воде. В одной стороны это соли, и как все соли должны быть растворимы в воде. Но растворимы – не значит смешиваются. В состав ионных жидкостей обычно входят алкильные хвосты – не очень большие, но и не очень маленькие: золотая середина это н-бутил. Это нам отчасти напоминает поведение простых спиртов – до пропилового они смешиваются с водой, н-бутиловый не смешивается, образуя отдельную фазу, при этом в нём достаточно велика растворимость воды. Так же ведут себя и ионные жидкости – они растворяют небольшие количества воды, чо помогает растворять в них простые соли, необходимые для той же реакции Судзуки-Мияуры, но не смешиваются в ней, по крайней мере, при комнатной температуре. При повышенной температуре от 50-60 градусов и выше многие ионные жидкости с водой смешиваются, образуя единую фазу. При охлаждении опять расслаиваются. Это очень удобное свойство. При комнатной температуре ИЖФ можно промывать водой, чтобы отмыть выделяющиеся при реакции соли, которые иначе будут мешать при рециклизации. Палладиевые комплексы остаются в ИЖФ.
Остающаяся после выделения продукта ионная жидкость с палладием может снова быть использована для кросс-сочетания. Заинтересованный наблюдатель может спросить – а в каком состоянии там палладий? Одно можно сказать точно – это не комплекс Pd(0). Это состояние точно не переживёт ни одну из трёх упомянутых манипуляций. Скорее всего, это какая-то форма Pd(2+), которая при следующей загрузке реагентов снова восстановится. Рано или поздно мы получим бесфосфиновую систему, так как фосфиновый лиганд тоже деградирует. В оригинальной статье делали двоекратную рециклизацию, что вполне объяснимо. Это тонкий момент в использовании ионных жидкостей для рециклизуемых систем. Количество рециклизаций будет ограничено – мы быстро утрачиваем понимание того, в каком состоянии там палладий. Скорее всего, система начинает вести себя как бесфосфиновая, благодаря тому, что часть палладия уходит в наночастицы, как обычно в таких случаях, а часть – в карбеновые комплексы. Чуть ниже разберёмся почему это скорее плохо, чем хорошо.
С этим пытались бороться с помощью специальных лигандов, которые, по замыслу, должны лучше держать палладий в ИЖФ. Марко Ломбардо с сотрудниками из Болоньи сделали фосфиновый лиганд, модифицированный хвостом с четвертичной аммонийной группой (M. Lombardo, M. Chiarucci and C. Trombini, Green Chem., 2009, 11, 574). Такой лиганд успешно держит палладий в ИЖФ. В этом месте удивленный читатель спросит – а что, разве трифенилфосфин не справлялся? Тонкий момент. Первые авторы утверждали, что палладий не уходит из ИЖФ, но не настаивали на том, что он удерживается там именно фосфином. Там что-то происходит – или наночастицы получаются, или он частично уходит в карбеновый комплекс, но больше двух рециклизаций делать не решились. В новом исследовании этой проблемой как раз озаботились. Для того, чтобы сделать систему более управляемой очень важно снизить температуру реакции. Монофосфиновые комплексы палладия не очень устойчивы, и равновесия диссоциации комплексов смещаются в сторону бесфосфинового комплекса, который дальше превращается в наночастицы и прочее. При 110ºС эти процессы более выражены, чем при более низких температурах. Исследователи из Болоньи тщательно оптимизировали условия и добились температуры 65ºС – это очень существенное смягчение. Реакцию они вели не в чистой ионной жидкости, а в ее смеси с водой (при этой температуре смесь становится гомогенной). И еще они взяли ионную жидкость, неимидазолийного типа и не дающую карбенов. В результате получили более реакционноспособную систему, спокойно выдержавшую 6 рециклизаций без снижения характеристик. Рециклизацию проводили сначала экстракцией продукта пентаном (надо сразу сказать, что это сойдёт только для самых несложных дифенилов, так как пентан – экстрагент неважный и забирает только неполярные продукты без серьёзных функциональных групп) и промывкой ИЖФ водой с последующей сушкой в вакууме (вопрос, зачем сушить в вакууме фазу, которую будут вновь смешивать с водой, остался без ответа – в Болонье слишком много искушений, чтобы тратить время на такие скучные мелочи).
Более-менее понятно, что ионные жидкости действиельно дают возможность строить фазо-разделяемые каталитические системы с рециклизуемым катализатором, и имеют в этом смысле определённые преимущества и достаточно уникальный набор свойств. Как растворители, или реакционные среды. Но остается вопрос, не участвуют ли они в реакции и более серьезно – как источники лиганда или ещё чего-нибудь существенного.
Источник этой идеи – еще одна знаменитая статья Вольфганга Херманна с пафосным названием Металлокомплексы NHC – новый структурный принцив в гомогенном катализе (Herrmann, W. A.; Elison, M.; Fischer, J.; Koecher, C.; Artus, G. R. J. Angew. Chem., Int. Ed. Engl. 1995, 34, 2371). Эта статья создает очень интересный казус – может ли ошибочное исследование стать важной вехой, положить начало новой области, и т.д. Кажется, что такого не может быть никогда, и ничего кромя пятна на репутации и насмешливых взглядов коллег – типа, поторопился, болезный, думать надо получше, да проверять результаты прежде чем статьи писать, – принести не может. Но вот ведь нет – наука, как и прочие области человеческой деятельности штука сложная, и из сора растут не только стихи “не ведая стыда”, но и новые научные направления. А когда уже выросли, кто спросит, что было в начале – была статья в мощном глянцевом журнале Ангевандте Хеми, и там что-то про имидазолиденовые карбены, что точно, никто уже не помнит, потому что есть уже пять тысяч статей про эти карбены, и во многих описаны безусловно выдающиеся достижения. Надо только понимать, что торопиться публиковать любую муть всё же торопиться не стоит, и не только потому, что не получится – что позволено Херманну, чрезвычайно авторитетному и могущественному в немецкой химии человеку, то не позволено вам, – но и потому, что из мути почти всегда рождается позор и другая муть, а исключения столь редки, что не стоит играть в эту игру. А Херманн тогда очень торопился заявить эпохальные открытия – он нацелился на пост ректора Мюнхенского технического университета, одного из самых мощных вузов в Германии, а значит и во всей Европе. И получил этот пост, и занимает его, кажется до сих пор. Торопился настолько, что пренебрег одним из важнейших неписаных правил западной научной этики – руководитель должен быть в конце списка авторов, а не в начале, а в начале должен быть тот, кто выполнил саму работу, аспирант или постдок. Впрочем, это не очень удивительно. Германия стала частью “запада” совсем недавно и не по своей воле, а до этого всегда считала себя особой цивилизацией, цивилизацией порядка и жертвенности, противостоящей меркантильному и подверженному всяким бунтам и революциям западу, под которым понимала Англию, Францию и их союзников. После нескольких практических попыток доказать величие и превосходство своей цивилизации кровью и железом, Германия потерпела чудовищный и беспрецедентный разгром, а ее остатки были вынуждены согласиться смирить гордыню и отныне играть по западным правилам, и это получилось настолько убедительно, что теперь Германия считается просто образцовой и едва ли не самой главной западной страной в Европе. Но, как видим, традиции остаются, и старинная привычка некоторых немцев к иерархии, чинопочитанию, большим и маленьким вождям и авторитетам никуда не делась.
Вернёмся к химии. В статье было описано, что простой комплекс палладия с самым простым имидазолиденом с двумя метилами превосходно катализирует реакцию Хека. Комплекс этот легко получается и проявляет просто совершенно невероятную устойчивость, что термическую, что химическую. И казалось совершенно очевидным, что и в катализе он будет сохранять свои карбеновые лиганды в совершенно неизменном виде – куда они денутся в довольно мягких условиях реакции (нагревание в растворе до 120-145 градусов, если его в твёрдом виде можно греть до 299 градусов (именно так в тексте статьи, не 300, а 299, видимо, для убедительности). Реакцию Хека взяли совершенно стандартную, ту самую, которую мы можем видеть в сотнях работ той самой гонки за TON, которую мы слегка затронули (за мной пока ещё более подробный рассказ об этой занятнейшей истории, тоже восходящей к статье Херманна, только другой).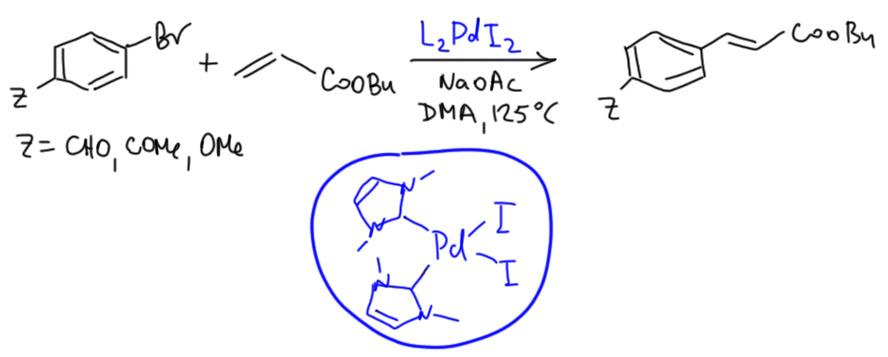
И что здесь не так? Только одно – никаких данных, кроме этой статьи, о том, что такой комплекс действительно работает как катализатор в реакции Хека нет. И в последующие 25 лет никто этот комплекс в практической работе с Хеком не применял. Только представьте такой невероятный случай – автор этого великого исследования только что возглавил крупнейший институт с практически неограниченными возможностями, и вроде бы исследования должны повалиться сотнями, но нет, ничего подобного не случилось. И не могло случиться, потому что опубликованные в статье данные однозначно говорят о том, что это – типичный очередной предкатализатор, который полностью разбирается в стадии предактивации и является источником бесфосфинового Pd(0), про который мы уже говорили, того самого, который работает через резервирование в наночастицах. Я напомню, что под термином “бесфосфиновый” я имею в виду палладий, не имеющий в координационной сфере координационно стабильных лигандов, классическим представителем которых являются фосфины. Да, в 1995 году этого еще никто не знал, но тогда надо было продолжать исследовать, определить, как положено в нормальных каталитических исследованиях, scope and limitations (возможности и ограничения), и т.п. И за последующие 25 лет NHC-комплексы не нашли никакого применения в Хеке. Это неудивительно, потому что мы знаем, что в Хеке с лигандами особенно не разбежишься, маленькие, типа вот этого карбена, занимают два места в мягких условиях, поэтому не работают, а в более жёстких просто уходят оттуда совсем, оставив палладий совсем без поддержки стабильных анциллярных лигандов.
В то же время, NHC-карбены нашли очень хорошее применение в кросс-сочетании, особенно в Бухвальде-Хартвиге, но и в Судзуки-Мияуре тоже, где они отлично помогают использовать менее реакционноспособные субстраты типа хлорпроизводных. Но – не все карбены, а только специфические, типа четвёрки IPr/SIPr/IMes/SIMes – очень большие, что было открыто в 1999 Стивеном Нолэном (J. Org. Chem. 1999, 64, 3804). Секрет их в том, что они не лезут к палладию больше чем по одиночке, и тем самым, почти идеально работают на очень активную форму катализатора с одним и только одним анциллярным лигандом в каталитической сфере. К тому же они очень донорные (но это не уникально, тот простой карбен, что у Херманна, тоже очень донорный, но их там два или и одного, а нужен только один). Карбены с простыми алкилами для серьёзного катализа оказались практически бесполезны.
Но образуются ли они вообще в условиях реакций Хека или кросс-сочетания при использовании ионных жидкостей. Видимо, да. Это, в общем, достаточно убедительно было показано в сравнении двух ионных жидкостей в реакции Хека (A. J. Carmichael, M. J. Earle, J. D. Holbrey, P. B. McCormac, K. R. Seddon Org. Lett. 1999, 1, 997). Никаких практически полезных протоколов из этой затеи не получилось, но был отмечен интересный факт – в имидазолильной ионной жидкости даже если палладиевая чернь начинает выпадать, в присутствии реагентов она быстро растворяется и более не выпадает. Это почти наверняка связано с тем, что карбен перехватывает палладий, скорее всего, на выходе из каталитического цикла, и резервирует его к карбеновом комплексе. Никаких особенных практических следствий из этого наблюдения нет, так как сам карбеновый комплекс всё равно не способен катализировать реакцию Хека, катализ идёт за счёт малой концентрации “бесфосфинового” палладия, а связывание излишков в карбеновый комплекс просто не даёт выпадать черни. И даже еще более убедительным свидетельством того, что карбеновые комплексы образуются из солей имидазолия, можно считать то, что уже в первом настоящем исследовании катализа карбеновыми комплексами в кросс-сочетании Стивен Нолэн предложил вместо свободных карбенов, которые все же не очень удобны так как плохо относятся к любой влаге, использовать соли имидазолия. Этот приём стал стандартным в реакциях с участием оснований. Как видим, карбоната цезия в довольно мягких условиях вполне хватает.
Резюмируем: карбеновые комплексы из имидазолиевых ионных жидкостей наверняка получаются, но это помеха, а не достоинство, потому что обычные алкильные, не имеющие объёмистых заместителей, карбены – плохие лиганды, и они скорее уводят палладий из каталитического цикла, а для реакций кросс-сочетания в мягких условиях это совсем плохо, а для реакции Мидзороки-Хека в жестких условиях, когда любой комплекс частично разлагается, выделяя активный (в этих условиях) бесфосфиновый палладий, это просто не имеет смысла, так как вместо карбена с тем же успехом можно взять картофельные очистки или банановую кожуру (если вы подумаете, что это тупая шутка, то нет, это описание совершенно конкретных работ, но об этом как-нибудь отдельно).
Регио- и стереоселективность реакции Мидзороки-Хека
Да, реакция Мидзороки-Хека радикально отличается от кросс-сочетания еще и в смысле селективности. Увы, эта реакция не обладает высокой регио- и стереоселективностью. В ранних работах на это не обращали особенно внимания, но по мере того, как реакция исследовалась, эти ее нехорошие свойства стали вылезать. Вообще, реакция МХ не очень хорошо работает в обычном межмолекулярном варианте:
- она очень избирательна к олефинам и практически используется только для терминальных олефинов, да и то далеко не для всех. Для дизамещенных олефинов она сильно ограничена, для тризамещенных почти совсем не годится, и есть буквально единичные работы с такими олефинами. Про тетразамещённые вообще говорить не приходится.
- она не обладает региоспецифичностью потому что ей не обладает ключевая стадия реакции – карбопалладирование. Направление карбопалладирования определяется кучей факторов, и электронных (в меньшей степени), и стерических (в большей), но ни один из этих фаторов нее работает со 100% определённостью
- стадия гидридного элиминирования тоже не стопроцентно надёжна. Жесткое требование син-геометрии не ограничивает альтернативы в реакциях терминальных олефинов, а кроме того, есть и вполне законные способы вовсе обойти это ограничение.
- ну и всё остальное, что мы уже и так знаем про трудности управления этой реакции современными анциллярными лигандами.
Реакция с терминальными монозамещенными олефинами порождает два вопроса – какой продукт получается, 1,1- или 1,2-замещенный олефин. 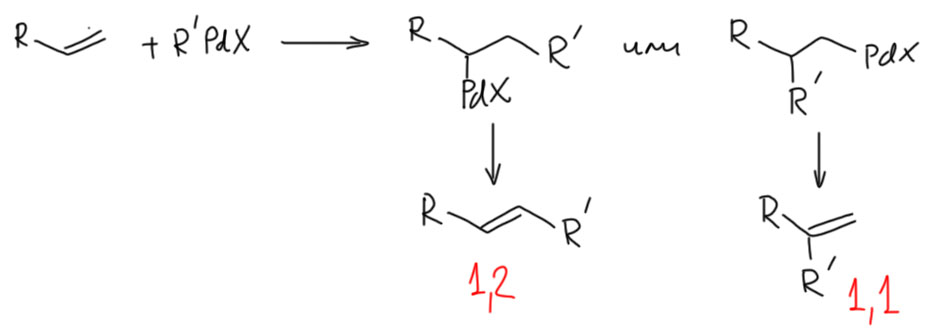 Обычно считается, что 1,2-, но это не совсем верно. Часто образуются смеси этих продуктов, а можно подобрать условия так, что в основном даже будет 1,1-продукт. Здесь скорее дело в том, что в практически важных случаях реакцию стараются вести так, чтобы получался 1,2-продукт. 1,1-продукт образуется в больших количествах, если используются бидентатные лиганды в так называемой полярной реакции Хека, когда для освобождния места в координационной сфере разными способами убирают анионный лиганд. Это очень особая реакция, и мы пока оставим ее в покое, тем более, что в реальном синтезе она используется гораздо реже, поэтому ей часто пренебрегают и даже забывают про это направление.
Обычно считается, что 1,2-, но это не совсем верно. Часто образуются смеси этих продуктов, а можно подобрать условия так, что в основном даже будет 1,1-продукт. Здесь скорее дело в том, что в практически важных случаях реакцию стараются вести так, чтобы получался 1,2-продукт. 1,1-продукт образуется в больших количествах, если используются бидентатные лиганды в так называемой полярной реакции Хека, когда для освобождния места в координационной сфере разными способами убирают анионный лиганд. Это очень особая реакция, и мы пока оставим ее в покое, тем более, что в реальном синтезе она используется гораздо реже, поэтому ей часто пренебрегают и даже забывают про это направление.
1,2-продукт образуется как основной в первую очередь с теми олефинами, которые особенно любят в реакции МХ – стиролами и акцепторными олефинами типа акцепторов Михаэля (акриловые кислоты и их производные и т.п.). Причина этого, скорее всего в том, что палладий взаимодействует с этими группами в процессе миграционного внедрения, и это снижает энергию активации именно на направлении, дающем 1,2-продукт. Вот, например, со стиролами, можно предположить взаимодействие дигапто-типа с ароматическим кольцом. Мы уже такое видели, например в некоторых лигандах Бухвальда, где такое взаимодействие держит координационное место, делая монодентатный фосфин временно бидентатным гемилабильным лигандом. Бухвальд, скорее всего, именно в реакции Хека “подсмотрел” этот эффект, превратив его в эффективный и эффектный приём дизайна новых фосфинов. В продукте карбопалладирования мы можем увидеть комплекс бензильного типа, формально сильно пожий на тригапто-аллильный, но значительно менее симметричный и намного более слабый – здесь он просто стабилизирует продукт карбопалладирования, ведущий к 1,2-продукту. Для другого излюбленного типа олефинов типа акриловых кислот и их производных ещё легче увидеть взаимодействие с неподелённой парой на гетероатоме. Обратите внимание, что ничего подобного не будет для второго аддукта (того, что привёл бы к 1,1-продукту), так как при син-карбопалладировании палладий подходит с противоположной стороны от заместителя, и сближения в переходном состоянии не будет. 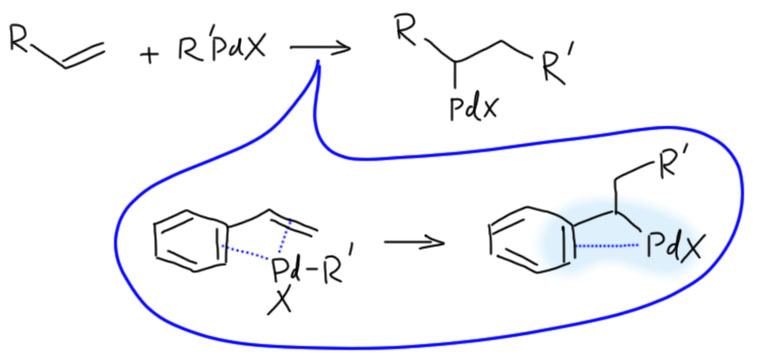
Теперь зададимся вопросом, что и почему образуется при 1,2-присоединении – транс- или цис-продукт. Это намного проще, потому что легко нарисовать оба пути и убедиться, что мы имеем дело с типичнейшим в органической химии явлением – кинетикой Кёртина-Гаммета. Напомню, что сей принцип гласит приблизительно следующее: если два разных продукта получаются из быстро взаимопревращающихся форм, то никого не волнует относительное содержание этих форм (их относительная устойчивость), а соотношение продуктов будет определяться кинетикой (константами скоростей) образования каждого из продуктов, или, что то же самое, но в других единицах измерения, разницей энергий переходных состояний. Вот мы и видим, что в переходных состояниях β-гидридного элиминирования, когда агостическое взаимодействие палладия и водорода фиксирует заслонённые конформации, в переходном состоянии, ведущем к цис-продукту стерическое отталкивание двух групп R и R’ дестабилизирует состояние, ведущее к цис-изомеру. В результате мы почти никогда не имеем более 5-8% цис-продукта, и даже часто просто не замечаем его, так как он просто теряется при выделении продукта реакции. Очень характерным исключением являются реакции акрилонитрила, в которых цис-продукта модет быть до 20% просто потому что циано-группа очень компактная. ![]()
Битва Мияуры с Судзукой
Я всё-таки настаиваю на написании “Судзуки” в соответствии с правилами передачи японских имён в русской графике (слог в романской графике “zu” передаётся как “дзу”, а не “зу”).
Здесь мы опять сталкиваемся с ситуацией, когда приходится решать, что важнее, эксперимент или объяснение, механизм. Эксперимент в химии всегда важнее. Сначала исследователь подбирает условия, позволяющие добиться желаемого эффекта, и только потом объясняет, почему так получилось. Мы иногда это путаем, потому что привыкаем читать и писать статьи, которые почти всегда начинаются с длинного введения, рассказывающего, как глубок был замысел, как точно было всё продумано, и как уверенно и прозорливо действовали учёные, конструировавшие новую реальность.
Всё это почти полная фигня. Исследование, если это действительно исследование, а не замена этила на пропил в уже известном, всегда риск неудачи. И неудачей обычно и кончается, за редкими исключениями, когда, переведя дух, уже можно садиться писать умную статью. Статья же не может рассказывать, как вы перепутали банки с реактивами, насыпали какой-то бессмысленной дряни, и вот – именно она оказалась ключом к решению.
Никто уже не расскажет, зачем в попытку устроить кросс-сочетание бромбензола с дибораном Норио Мияура добавил такое странное основание, как фенолят, очень слабое.
Задача была более-менее стандартной – устроить классическое кросс-сочетание, но так чтобы получить связь C-B. Но для этого борный остаток должен был стать нуклеофилом, чтобы въехать в координационную сферу палладия в переметаллировании. Безусловно, не с совсем прямом смысле этого слова, но хотя бы в формальном, потому что борный остаток должен переехать на палладий, заменив на нём нормальный X-лиганд, то есть став на нём X-лигандом. А бор вообще как, по сравнению с палладием, что у него с электроотрицательностью? Если смотреть на атомные электроотрицательности, то палладий однозначно более электроотрицательный элемент в этой паре, хотя разница невелика. Строго говоря, это не так важно – электроотрицательность всё же величина веьма абстрактная, она хорошо работает, когда разница велика, и становится почти произвольной, когда мала. В этом случае хорошо было бы руководствоваться не атомной электроотрицательностью, а какой-то эффективной величиной, учитывающей эффекты заместителей, гибридизацию и пр. Такими величинами в прошлом веке много занимались, пытаясь создать универсальные шкалы, но в конце концов поняли, что произвола в таких шкалах гораздо больше, чем в известной всем со школы обычной электроотрицательностью (совершенно не важно, в какой формулировке). Поэтому, хотя это уже довольно смелый шаг в сторону от уже устойвшейся схемы кросс-сочетания, можно было хотя бы на эту тему не париться, а считать, что раз на боре висят два кислорода, его эффективная электроотрицательность становится выше, и он будет вести себя по отношению к палладию как настоящий неметалл. Можно ожидать переметаллирования, иными словами. Но вот что взять вместе с бором? По классической схеме это должно быть что-то типа связи B-Sn или B-Zn. Такие соединения (бориды) есть, но даже если бы с ними что-то и получилось бы, желающих применять такое не нашлось бы почти точно. А задача была не просто сделать какую-то реакцию, а создать удобный и гибкий метод, который заинтересует настоящих синтетиков. И тогда взгляд упал на соединения со связью B-B, некоторые из которых были вполне доступны и не являлись экзотикой. Это не то, что в те времена стояло на каждой полке, но это вполне устойчивые вещества, которые можно было, при успехе, предложить компаниям, производящим реактивы, так, чтобы это можно было купить, а не делать.
Осталось сделать связь B-B несимметричной, пригодной для переметаллирования, чтобы один бор взял пару электронов, а второй отдал. В химии это вполне стандартный процесс – разве не так ведут себя в реакциях молекулы галогенов, или соединения со связями кислород-кислород, кислород-галоген, кислород-азот, азот-галоген и пр. Но просто в молекуле любого диборана так не получится, потому что у бора уже только 6 электронов, и отдавать еще пару – это перебор. Первоначальный замысел, скорее всего, и состоял в том, чтобы добиться координации жёсткого основания Льюиса по одному из атомов, и в такой молекуле атомы бора уже автоматически получат нужные роли, а та часть, в которой бор остался трёхкоординированным формально станет нуклеофилом. Конечно, это типичная авантюра – сделать нуклеофилом, хотя бы формально, атом с 6 электронами! 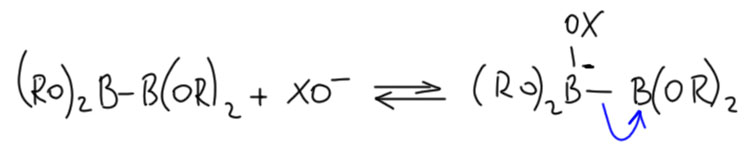
Но в этом месте поджидает засада. Если такая реакция и пойдёт, то образующийся боронат в тех же условиях может сразу же вступисть в реакцию Судзуки, потому что в этот момент в реакционной смеси много электрофила – большой избыток по сравнению с только начавшим получаться боронатом.
В этом месте можно понадеяться на то, что боронат не так легко реагирует в C-C кросс-сочетании, как бороновая кислота. Тогда у нас возникла бы некоторая фора, которая позволила бы продвинуться в C-B кросс-сочетании раньше чем в дело вступит C-C. И чем больше разница, тем больше будет фора. На самом деле, именно этим объясняется, почему в борилировании пошли не простые (метиловые и т.п.) эфиры диборной кислоты, а циклические, да и не просто с этиленгликолем, а с рогатыми диолами – пинаконом или, позднее, неопентилгликолем. Такие эфиры действительно сильно уступают бороновым кислотам в скорости C-C кросс-сочетании в сравнимых условиях, и они весьма устойчивы к “переэтерификации”. Это оказалось очень хорошим выбором.
Вопросы 2021 года
Какие ещё металлы применяются в кросс-сочетании кроме палладия и никеля
Очень интересный вопрос, хотя в нём заложена небольшое недоразумение. Палладий это официально драгоценный металл, такие металлы котируются на мировых биржах в тройских унциях, то есть очень маленькими порциями. Палладий – один из самых дорогих металлов, по цене уступающий только родию, который за последние 10 лет из-за интенсивного применения в катализе ушел в отрыв и один стоит больше, чем все остальные драгметаллы, включая золото, платину и проч. Никель и медь – это обыкновенные цветные металлы, которые котируются тоннами. Никакого сравнения не может быть в цене палладия, с одной стороны, и никели или меди, с другой. Замена палладия на никель уже дает настолько гигантский экономический эффект (это приблизительно три порядка в цене сырья), что дальше, в принципе, говорить не о чем и в дело должны вступать другие соображения. Про серебро мы не говорим, у серебра до сих пор настолько узкая роль в катализе, что пока говорить не о чем.
Медь действительно немного дешевле никеля. По котировкам основной мировой биржи металлов – LME (London metal exchange) в прошлом году средняя цена никеля была около 16 тыс долларов за тонну, меди – около 8 тыс долларов за тонну, хотя все эти цены год от года сильно изменяются в зависимости от мировой конъюнктуры. Для применения в катализе эта разница совсем неощутима.
Что касается химии, то в современной химии в кросс-сочетании царят три металла – палладий на первом месте вне конкуренции, потому что только катализаторы на основе этого металла обеспечивают весь колоссальный диапазон реакций и методов, они наиболее надежны, эффективны, удобны в работе – в общем, незаменимы, хотя и чертовски дороги.
На втором месте не никель, а именно медь. Но медь очень хороша в кросс-сочетании с образованием связей угоерод-гетероатом (C-N, C-O, C-S), где она довольно успешно конкурирует с палладием. Для кросс-сочетания углерод-углерод медь до сих пор имеет нишевое применение, хотя и очень важное, это сочетание алкил-алкил, где палладий просто позорно проваливается.
Никель – тоже металл для отдельных задач, не претендующий на широту. Он по прежнему очень широко применяется для реакции Кумады, где успешно держит палладий на втором месте, но в остальных реакциях кросс-сочетания не завоевал доверия синтетиков по разным причинам. С другой стороны, у никеля есть особая роль – из-за невероятной мощи в окислительном присоединении никелем можно раскачать многие субстраты, нереакционноспособные с палладием. Об этом тоже будет сказано в шестой аудио-лекции.
В современной химии в кросс-сочетании очень ярко проявили себя соседи никеля – кобальт и особенно железо. Кобальт был первым металлом к кросс-сочетании, открытым еще Харашем в 1941, но с тех пор ушел из внимания и долго был забыт. В последние 10 лет за него взялись основательно – результаты хороши, хотя пока о широком применении говорить рано. Еще более интересно железо. Возможности простых комплексов железа(3+) катализировать кросс-сочетание галогенпроизводных с гриньярами открыл прямо перед реакцией Кумады-Тамао-Коррью выдающийся американский химик Джей Кочи. Но поскольку началась эпоха палладия, про эти работы забыли на почти 40 лет. Но в 2010-х за железо зацепились, принялись его мощно исследовать, и буквально за несколько лет появилась новая область в кросс-сочетании, необычайно богатая на удивительные фокусы. У железа, в отличие от палладия, доступен целый ряд окислительных состояний – -2, 0, +1, +2, +3. Это делает механизмы намного более сложными для распутывания, но и возможностей там очень много – новых достижений можно ждать каждый день. На будущий год включу железное и кобальтовое кросс-сочетание в курс.
Кросс-сочетание - это гомогенный или гетерогенный катализ?
Еще один с виду банальный вопрос, простой и очевидный ответ на который – конечно, гомогенный – не совсем точно соотвествует действительности.
Сразу скажем, что кросс-сочетание здесь – просто архетип, самый типичный и важный представитель всего обширного множества реакций, катализируемых комплексами переходных металлов, являющихся предметом настоящего курса. Поэтому можно так и переформулировать: Что мы изучаем в этом курсе, гомогенные реакции или гетерогенные?
С самого возникновения этой области исследований основная задача формулировалась вполне явно и недвусмысленно – развитие именно гомогенного катализа, даже точнее, катализа в жидких средах, в растворах, хотя это и довольно очевидно – попробуйте представить себе гомогенный катализ в газовой фазе (то есть, и катализатор тоже должен быть газообразным!) или твердой фазе – в этом случае реакционная смесь должна была бы быть или твердым раствором, или аморфной – но последнее в любом случае двусмысленно, так как нет принципиальной разницы между жидкостью и аморфным телом.
С самого начала гомогенный катализ именно противопоставлялся гетерогенному, который уже был отлично разработан и применялся в огромном количестве промышленных процессов в основном в вариантах: твердый катализатор – реагенты в газовой фазе или растворе. Гетерогенный катализ очень хорошо тем, что катализатор легко отделяется от реакционнй смеси и продуктов реакции, что радикально упрощает выделение продуктов и регенерацию катализатора, снимает проблемы с дороговизной некоторых катализаторов, потому что они не теряются, а время от времени регенерируются без затрат на новые количества саого ценного компонента – металла. Но за полвека интенсивной разрабоки и эксплуатации гетерогенных каталитических систем к ним возникли большие претензии – обычно это не очень высокая селективность, почти полная невозможность разработки стереоселективных вариантов, и, самое главное, из чего проистекает все остальное – в принципиальной невозможности хорошо разобраться в механизме, а значит и в осмысленной разработке – трудно рационально заниматься тем, что мы не понимаем и понять не можем – весь 20 век исследования поведения молекул на поверхности твердых материалов далеко не ушли из-за отсутствия методов исследования. Если вы когда-нибудь видели статьи по исследованию гетерогенного катализа с попытками осмыслить механизм, не могли не поразиться совершенно наивным картинкам – какие-то шарики куда-то там цепляются, сидят на каких-то лесенках и решёточках, и что-то натужно изображают – настолько это контрастирует с тем, как мы привыкли рисовать и понимать настоящие механизмы реакций. Но ничего другого там и не нарисуешь, настолько поведение молекул и атомов на поверхности отлично от нашего понимания поведения тех же молекул в растворах. Только в нашем веке в этом смысле наметились серьёзные сдвиги.
Поэтому пошел гомогенный катализ. С самого начала это было просто райское ощущение – наконец-то исследователь может нарисовать нормальный механизм по всем правилам структурной химии, разделять многостадийный процесс на элементарные реакции, пытаться изучать их по отдельности, выделять или ловить интермедиаты, измерять кинетику и всё такое – уровень понимания процессов сразу уехал очень далеко, тем более что к этому времени огромные успехи сделали и координационная, и органическая химия. Появилась возможность целенаправленно конструировать катализаторы, и с ними идти на штурм крепостей, которые не сдавались – в первую очередь, энантиоселективных реакций. Я очень хорошо помню, что в 1970-х оптическая чистота продукта (энантиомерный избыток) в 5% считался великим достижением, и на полном серьёзе доказывалось, что большей чистоты принципиально добиться никогда не получится. А через 10 лет 95% перестали считаться чем-то необычным – и ключом оказались те самые гомогенные катализаторы и правильно подобранные хиральные анциллярные лиганды. В результате всего за несколько десятилетий мы получили мощнейшую новую химию, которую в этом курсе и пытаемся хоть немного освоить.
Но, как всегда бывает, никакое решение не становится абсолютным. Быстро стало понятно, что за новые достижения приходится очень дорого платить – катализаторы оказались безумно дороги и из-за платиновых металлов во многих, и из-за специальных и очень дорогих лигандов, и из-за непривычной для практически применимой химии чрезвычайной тщательности и высоким требованиям к чистоте всего и самой технике выполнения экспериментов и синтезов. Стало очень трудно отделять катализатор от продуктов, а следовательно и регенерировать его, и не только регенерировать, но и банально очищать продукт от примеси соединений тяжелых металлов, а это неприемлемо, если синтезе делается не просто так, а для производства, например, чего-то, используемого в медицине. Проблема оказалась не менее острой, чем старые проблемы недостатка селективности.
И вот тогда возникло желание как-то сшить гомогенный и гетерогенный катализ, желательно сохранив преимущества обоих, возник запрос на гетерогенизацию гомогенного катализа. Задачу стали с самого начала решать иммобилизацией гомогенных катализаторов на твердых подложках (сначала просто полимерах, а потом в ход пошли разнообразные современные материалы, которых тоже становится все больше и больше). Такие системы работали, но без блеска, свойственного настоящим гомогенно-каталитическим реакциям, прежде всего потому, что соединить иммобилизуемость и собственно эффективность лигандов и комплексов оказалось намного труднее ожидаемого.
Дальше возникла химия наночастиц, возникла гипотеза, что наночастицы переходных металлов могут быть особенно эффективны в качесве катализаторов, а за счет модификации поверхностных атомов лигандами можно добиться и желаемой модуляции свойств и селективности. Тут же возник вопрос – а наночастица это отдельная фаза или просто очень большая молекула (кластер) в растворе. То есть, это гомогенный катализ или гетерогенный? Ответа на этот вопрос нет, потому что это не сущностный вопрос, а скорее, философский, а значит – каждый волен на него отвечать, как хочет.
Дальше выяснилось, что наночастицы тоже надо как-то отделять и регенерировать. Возникли иммобилизованные наночастицы, и это уже совсем непонятно что с точки зрения разделения фаз.
Параллельно стал развиваться фазо-разделяемый катализ – это когда реакция идет в одной фазе (она гомогенная), но после реакции осуществяется разделение фаз, при котором катализатор оказывается в одной, а продукты в другой. И пока такие системы разрабатывали, выяснилось, что и там все не так просто, и что разделение фаз во многих случаях все же происходит во время реакции, а не после нее.
В наше время все стало совсем запутанно. Возникло даже такое направление, как катализ индивидуальными молекулами на поверхности (single-atom catalysis), соединяющий преимущества понимания механизма реакции с помощью известных каталитических частиц, с обычной гетерогенностью – реакции идут на поверхности, но там уже не глупые шарики, а конкретные комплексы и все остальное, как мы любим – каталитический цикл, вход, выход, и т.д. Это очень свежая область, немного обескураживающая громогласностью задач, изощренностью экспериментальных методов, и отсутствием ответов на простые вопросы, например, о каталитической эффективности таких систем. Но – интересно, даже захватывающе.
А вот и совсем простой вопрос по поводу гомогенности или гетерогенности. Как мы знаем, классическая реакция Судзуки-Мияуры идет в органическом растворителе, к которому добавляют немного водного раствора щелочи или чего-то типа этого. И этот раствор не смешивается, остается отдельной фазой. Вопрос – а где идет сама реакция кросс-сочетания? Если палладиевые комплексы в органичекой фазе, субстраты там же, а совершенно необходимый для активации переметаллирования гидроксид – в одной. Как-то тут тоже что-то важное происходит гетерогенно, скорее всего в режиме фазового переноса, в котором межфазным переносчиком служат комплексы палладия.
В общем, получается, что однозначного ответа на простой вопрос нет, и это просто проблема неадекватности простого разделения гомогенный-гетерогенный для сложной системы явлений, вовлеченных в течение реакций с участием комплексов переходных металлов в катализе.
Поэтому не будем придираться к словам – и займемся вместо этого изучением сути.
Бесполезна ли чернь, палладиевая чернь
Да, чернь очень часто вываливается в реакциях с участием комплексов палладия. Вываливается – очень точное слово, это действительно часто происходит неожиданно – пару секуд назад был прозрачный раствор, все было хорошо – и вдруг, раз – и всё черное, и еще через несколько минут на дне валяются черные крупинки.
Мы знаем, что нульвалентный палладий не обязательно валится в осадок в виде черни. Он не валится в двух случаях:
- Pd(0) стабилизируется хорошими лигандами, в первую очередь, фосфиновыми, но, возможно, и какими-то другими. Такой палладий может очень долго быть в растворе, теоретически бесконечно, но это только в том случае, если раствор тщательно изолирован от доступа воздуха – кислород медленно, но верно разрушает лиганды и быстро или медленно оставит палладий без защиты.
- Pd(0) участвует в каталитической реакции, причём скорость стадии с участием этой формы палладия больше скорости слипания атомов палладия – это очень важный случай, разобранный в одной из лекций под рубрикой “бесфосфиновый катализ”
И в том, и в другом случае палладий может долго оставаться в растворе, но в конце концов, вывалится – или реакция замедлится, или лиганды разрушатся. Те, кто реально делал реакции кросс-сочетания, очень хорошо это знают – вываливание черни почти всегда происходит рано или поздно и означает либо, что реакция закончилась, субстраты израсходовались, и в этом случае палладий, оставшийся без дела очень часто берется за свои собственные лиганды и уничтожает их, оставаясь без поддержки. Либо, что хуже, если реакция идет не очень хорошо, что-то пошло не так, воздуха напустили или растворители грязные или еще что-то, но тогда реакция на этом заканчивается задолго до полного расходования субстратов. Нужно проанализировать ситуацию, найти причины и принять меры.
Как только чернь вывалилась, реакции останавливаются. Чернь – это беспорядочные сростки более-менее крупных частиц палладия, переросших стадию наночастиц, слипшихся в слабые агрегаты и ставшие слишком тяжелыми для того, чтобы держаться в растворе за счет обычного теплового движения. Иногда чернь выпадает на стенках посуды, особенно когда стекло старое, изношенное, сильно потертое мешалками – частицам палладия есть за что зацепиться. Иногда на мешалках, опять таки, когда они старые, потертые, с окисленной поверхностью.
Может ли чернь участвовать в реакциях кросс-сочетания или похожих. Да, может. Частички черни – это не кристаллы металла с правильной поверхностью, а реально сростки субмикро- и больших наночастиц, там очень сложный и нерегулярный состав, и много мест, где поверхность огромна, атомы палладия торчат во все стороны во всяких дефектах и прочих наноразмерных неровностях. Такой палладий может взаимодействовать с реакционноспособными субстратами, прежде всего иодпроизводными, но и бромпроизводными с акцепторными группами (любимый субстрат всех исследователей, которые хотят доказать, что кросс-сочетание – такая реакция, которую можно провести со всем чем угодно, включая битый кирпич или картофельные очистки – пара-бромацетофенон, обладающий совершенно чудовищной реакционной способностью в окислительном присоединении, он способен подхватить миллионную часть палладия и даже меньше, то есть то, что находится н уровне почти неопределимых следов; во всех таких исследованиях оказывается, что реакцию вели в той же колбе или с той же мешалкой. что и нормальную реакцию кросс-сочетания днем, месяцем, или годом ранее). Окислительное присоединение уносит часть палладия в раствор и начинается обычная бесфосфиновая реакция в растворе.
Такими вещами увлекались многие лет 25 тому назад, потом это всем надоело, потому что предсказать результат таких реакций очень сложно – образцы черни, выпадающие в разных экспериментах отличаются друг от друга совершенно случайным образом, и количество случайно доступного палладия может быть совершенно разным в двух параллельных и с виду идентичных опытах. Выходы и время реакции в таких экспериментах непредсказуемы. Участвуют в таких реакциях только избранные субстраты типа того же п-бромацетофенона или иодбензола, и любые попытки распространить реакцию на что-то реально нужное проваливаются. Поэтому эта химия сама собой заглохла практически без следа, хотя иногда бывают курьёзные рецидивы. Это, кстати, поучительно – никогда не занимайтесь сомнительной фигнёй – даже если получится накропать статейку в какой-нибудь невзыскательный журнальчик, проку от нее не будет, и в лучшем случае она будет забыта.
С другой стороны, чернь очень удобна в том смысле, что ее легко отделить от реакционной смеси, она обычно очень легко отфильтровывается, причем не надо ее соскребать с фильтра – используйте один фильтр на много реакций, и когда сильно почернеет, просто промойте его царской водкой. А из раствора уже достанете палладий, который очень легко перевести обратно в ацетат или хлорид. Палладий на дороге не валяется, нынче он дороже золота и платины.
Как отличить комплекс с дигаптолигандом от трёхчленного металлацикла
На этот вопрос частично ответ есть на страничке про металлациклопропаны. И еще будет в аудио-лекции про олефины.
Back-donation делает из олефина металлациклопропан. А с аллильным лигандом может произойти что-то подобное?
Вопрос крайне непростой и очень интересный. Вообще-то аллильные лиганды обычно считаются невосприимчивыми к эффектам back-donation, и в общем легко понять почему. В отличие от нейтральных лигандов типа олефинов или диенов, которые с удовольствием берут лишние электроны от сильно-донорных низковалентных металлов, аллильный лиганд уже формально четырёхэлектронный, он уже имеет аллильную систему, заселённую четырьмя электронами. Брать еще электроны этой системе не очень хочется, так как это создаст откровенно избыточную плотность на небольшом лиганде. Это можно сформулировать как низкую пи-кислотность аллильных лигандов. То же самое справедливо для циклопентадиенильной системы.
Поэтому аллильные комплексы в подавляющем большинстве случаев очень просто устроены, представляют собой или η1 или η3-комплексы, а металл обычно находится в валентном состоянии, не предполагающем большого желания устроить хороший back-donation. Иногда бывают мостиковые аллильные комплексы, в которых один металл хватается за один углерод, а второй – за двойную связь, как например, в таком комплексе платины (W. S. McDonald, B. E. Mann, G. Raper, B. L. Shaw, G. Shaw J. Chem. Soc. Chem. Commun., 1969, 1254) Но и тут платина самая обычная, плоскоквадратная. 
У ранних переходных металлов аллильные комплексы встречаются часто, но и там не представляют ничего необычного, и металл в них обычно сразу имеет конфигурацию d0, так что отдавать больше нечего.
Попробуем представить, что было бы, если бы аллил всё же решил разжиться лишними электронами. Безо всяких МО это можно вполне разумно представить просто переносом электрона. Сначала вспомним, что происходит с олефином. Можно перенести один электрон или два – результат будет один и тот же (потому что это не настоящий перенос, а именно что мысленный, как говорят религиозные люди о всяких мистических материях или молитвах, умнóй, т.е. происходящий в уме).
Как видим, разницы нет никакой, потому что ковалентная связь все равно в расчете степени окисления даст то же самое. Итого металл увеличивает формальную степень окисления на 2 единицы.
А теперь попробуем проделать то же для аллила. Исходный аллил изобразим аллильным анионом, в полном соответствии с тем, как мы его считаем в координационной сфере. Переносим электрон – и получаем вроде вторую связь, но и какой-то странный радикал на среднем атоме. 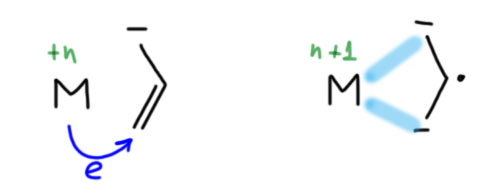 Что с ним делать? Чёрт знает. Понятно только, что просто не получается. Давайте что ли второй электрон перенесём, а то тут совсем тоска.
Что с ним делать? Чёрт знает. Понятно только, что просто не получается. Давайте что ли второй электрон перенесём, а то тут совсем тоска. 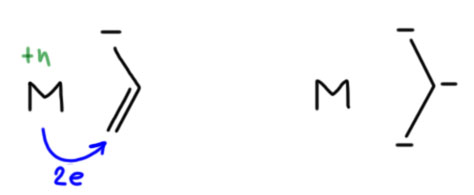
Ну, это совсем ни в какие ворота не лезет – трианион какой-то. Что-то другое с этим сделать не получится, потому что у аллильной системы всего 3 орбитали, вот их можно все занять, тогда на всех орбиталях будет по паре электронов. В общем, понятно, что аллильная система не очень приспособлена для back-donation.
В этом месте можно было бы всё бросить и успокоиться. Но, как ни странно, в этом странном трианионе есть некоторый смысл. Только проявиться он имеет шанс не в комплексе с одним металлом. А вот если с двумя – в двухядерном комплексе, чтобы было рядом два металла, каждый впрыснул бы в аллил по одному электрону, и тогда такой лиганд мог бы стать мостиковым, взяв каждый из металлов дигапто-способом. Получится что-то такое: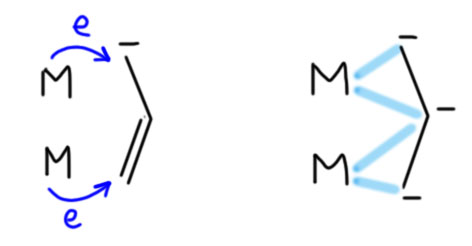
Эх-хех… Выглядит малость завирально, но – как ни странно – нечто подобное как раз существует, было обнаружено Фрэнком Коттоном в 1970-х. Такие комплексы дают, например, металлы 6 группы, чрезвычайно склонные к образованию двухядерных и многоядерных комплексов в промежуточных степенях окисления. Все три металла группы дают практически одинаковые по структуре димеры бис-аллильных комплексов (быстренько в уме считаем – мономер имел бы степень окисления +2, а это для раннего переходного металла явная заявка на back-donation, и счёт электронов всего 12 – высокую степень ненасыщенности. Двухвалентные металлы 6-й группы знамениты своей способностью насыщаться с помощью кратных связей металл-металл, для валентного состояния +2 это может быть до четверной связи (четыре оставшиеся d-электрона могут распарится – пустых орбиталей для этого достаточно – и зацепиться друг за друга. Не будем подробнее про связи металл-металл, пока у нас их в курсе нет, но я уже чувствую потребность их туда включить, потому что вижу, как в последние годы в катализ пошли многоядерные комплексы, пора. Включу на будущий год. Пока ограничимся таким куцым объяснением.
Так вот, в этих комплексах два аллила обычные, а два связаны именно так – это мостик, в котором каждый металл имеет дигапто-связь с аллилом. А почему мы считаем так, а не как выше в комплексе платины – мостик с разной гаптностью? Структура! Там она несимметрична, а здесь совершенно симметрична. А значит оба металла связаны с аллилом о-ди-на-ко-во!
Как же считать такие комплексы? Это очередной пример неопределенности. Само образование такого комплекса, как мы видим, предполагает серьёзную роль back-donation. Но в картинке мы этого не учитываем – мы рисуем именно димер мономерного бис-аллильного комплекса, в котором металл имеет степень окисления +2, и именно это дает при димеризации четверную связь. Но если бы мы перенесли полностью один электрон в back-donation, степнь окисления увеличилась бы до +3, а это предполагает только тройную связь. Так какая там связь?! Неужели это непонятно?! Строго говоря, не очень. На связи не написано. Кратность связи обычно оценивают по расстоянию между атомами – чем связь короче, тем больше порядок. Нужно иметь приличную кучу комплексов с разными связями, чтобы наьрать статистику, причем в этом наборе должны быть более однозначные структуры. Но эта оценка тоже не дает однозначного ответа, потому что диапазоны изменения длин связей немаленькие. Можно дополнить это хорошим расчетом, но и там есть место сомнениям. В общем, это не так важно – приходится привыкать, что хотя все характеристики комплексов работают очень хорошо, но есть некоторое количество более сложных структур, для которых остается некоторая неопределенность. Нарисуем с четверной и Mo(2+).
Так что комплексы такие всё же сущестуют, хотя это очень большая редкость, и с тех пор примеров почти не прибавилось. Сделаем вывод, что аллил действительно плохо приспособлен для сильного back-donation, но если металл сильно попросит, то всё же может иногда изобразить что-то похожее.
Может ли стерика помешать достижению правильной геометрии комплексов
Могут ли объемные заместители в исходном алкене (а также объемные L) препятствовать изменению геометрии комплекса в плоско-квадратную?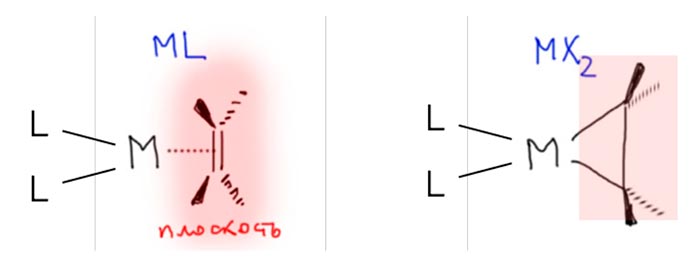
Здесь не всё однозначно, хотя бы потому что комплексы такого типа всё же более характерны для ранних переходных металлов в промежуточных степенях окисления, а плоский квадрат – это скорее о поздних переходных металлах и 16-электронных комплексах, в которых одна p-орбиталь выключена и находится в резерве для ловли аксиальных лигандов. У поздних переходных металлов тоже бывают комплексы с олефинами и ацетиленами с высокой долей back-donation и тенденцией к изменению степени окисления. Но проблема в том, что у η2-комплексов олефинов не очень точно задана геометрия, так как олефиновый фрагмент имеет возможность вращения, и один чёрт разберёт, какой геометрии комплекса лучше соответствует структура – квадратной или тетраэдрической. Когда же проявляется back-donation, конфигурация фиксируется более надёжно из-за регибридизации, и да – это хорошо видно на структурах. Немного позже постараюсь подобрать структуры, которые показывают это достаточно ясно, структур таких комплексов наделано довольно много, так как олефиновые комплексы легко получаются и довольно часто всплывают в разных исследованиях.
Что же касается стерики, то тут не очень легко подобрать ситуации, в которых это могло бы проявиться. Стерика вообще очень важна в химии переходных металлов. Часто она просто препятствует образованию комплексов. Например, сильно-замещённые олефины вообще весьма редко образуют устойчивые комплексы. Это очень ярко проявляется во многих реакциях – уже трёхзамещённые олефины весьма неохотно реагируют и в гидрировании и в других реакциях присоединения, в которых должны образовываться олефиновые комплексы. А уж четырехзамещенные олефины вообще почти всегда совершенно нереакционноспособны. Химия – наука экспериментальная, и далеко не всё, что мы можем нарисовать на бумаге, реально образуется и может быть исследовано.
Можно ли сделать что-то подобное реакции Кулинковича с циклами большего размера
Но сразу возникает вопрос, только ли циклопропаны можно собирать таким образом? Ведь если мы увеличим цепь на один атом (понятно, что реакционные центры должны быть сближены, поэтому сразу отброшена идея о сборке больших циклов таким образом), открывается путь для синтеза циклобутановых фрагментов в ТМ.
Всё не так просто. В химии переходных металлов реакций не меньше, чем в обычной органике. а гораздо больше. Но далеко не все из них заслуживают статуса именной реакции или укоренившегося метода. Для этого мало просто реагировать и давать что-то интересное – уж интересного-то в химии переходных металлов нет числа. Нужно решать какую-то важную задачу в синтезе, иметь возможности для дальнейшего развития, делать вещи изящно и гибко, или наоборот предельно тупо, но эффективно и просто. Вот реакция Кулинковича в полной мере соответствует этим требованиям – с её помощью довольно просто и быстро получаются функционально-замещённые циклопропаны, а это весьма востребованные соединения. Она изящна, потому что решает важную проблему необычным, но очень эффективным способом, хорошо поддается развитию, недаром за неё быстро уцепились два чрезвычайно плодовитых химика, немец Армин де Мейере и канадец Виктор Снечкус, и стали штамповать работу за работой, наворачивая на идею Кулинковича новые подходы и стереоселективность.
Металлациклов с ранними переходными металлами, и четырёх-, и пяти- и шести- и так-далее-членных, с теми же титаном и цирконием очень много. Они охотно образуются в самых разных процессах, от всяких аналогов метатезиса, до циклоолигомеризации и т.п. У них разнообразная химия, из них много что получается, и да, они реагируют с органическими электрофилами практически так же, как титанациклопропаны Кулинковича. Но реакции почти всегда стехиометрические, хотя бы потому что циклы большего размера вполне стабильны и сами по себе в конечные продукты не превращаются, их надо разлагать протолизом или еще как-то. Поэтому из них не получается удобной реакции, да и продукты, к которым они приводят можно получить десятками других способов.
Вот например, титанациклобутены, образующиеся из титановых карбенов Шрока и ацетиленов, отлично реагируют с нитрилами, ровно так же, как в Кулинковиче реагируют сложные эфиры (Doxsee, K. M., Garner, L. C., Juliette, J. J. ., Mouser, J. K. ., Weakley, T. J. ., Hope, H. Tetrahedron, 1995, 51, 4321). Может войти и один и два нитрила по каждой из связей Ti-C, ровно как и должен карбанион присоединяться к нитрильной группе. Продукты – шестичленный титанацикл и восьмичленный хелат приходится гидролизовать солянкой. Получаются вполне полезные вещи, но так их получать не будет никто и никогда. Что и отражается в довольно скромном цитировании этих работ, и полном отсутствии продолжения и развития. 
А вот достижение посвежее, очередное расширение реакции Кулинковича, но с получением 4-членного цикла, правда не карбоцикла, а гетероцикла, азетидина ( L.Kürti et al Angew Chem Int Ed 2019, 58, 14219). Титанациклопропану подсунули не сложный эфир и не амид, а оксим, точнее его О-эфир. Такие производные оксимов интересны тем, что азот в них не нуклеофилен, а электрофилен благодаря наличию уходящей группы. Поэтому после первого присоединения, происходит атака не на атом углерода с уходящей группой, а на атом азота. 
Эта работа Ласло Кюрти и сотр. довольно ясно показывает, что у реакции Кулинковича впереди ещё много интересного. Возможно, и циклобутаны не за горами, надо только подобрать правильный реагент для внедрения в титанациклопропан.
Экономия атомов в реакции Кулинковича
Зачем в модификации реакции Кулинковича нужен CyMgBr? Почему, нельзя взять смесь реактивов Гриньяра, где один из них метильный, а другой – в будущем нужный лиганд-актор (1:1)?
Это два разных вопроса. Циклогексильный (или циклопентильный) гриньяры берут в методе подмены олефина (об этом рассказано в аудио-лекции). Вторая проблема в оригинальной реакции Кулинковича – бесполезный расход одного из гриньяров, который работает только как основание во внутрисферном элиминировании протона. В начале это всегда были этилы, и на этилы всем плевать, невелик расход. Но когда научились использовать более сложные гриньяры, бесполезный расход одного эквивалента стал обузой. В современном синтезе такие вещи очень плохо влспринимаются, так как портят экономию атомов. Реакция Кулинковича и так не является эталоном экономии атомов из-за низкой каталитической эффективности, но так как алкоксиды титана действительно очень дешевы, это не так страшно. А вот превращение одного эквивалента более сложного гриньяра в отходы нехорошо.
С этим довольно быстро научились справляться именно так – сначала алкоксид титана обрабатывают эквивалентом метильного гриньяра, и только потом вводят гриньяр для задней стенки циклопропанола. С некоторого времени этот подход стал настолько стандартным, что первую стадию в схемах реакций стали опускать, и прямо пишут в качестве реагента метилтитан. И в методе подмены олефина делают точно так же – сначала метильный гриньяр, потом циклогексильный или циклопентильный, и потом собственно олефин.
У этого приёма, весьма популярного в синтезе, впрочем есть своя цена – он совсем не приспособлен к каталитическому варианту. Просто представьте себе, как это будет, если алкоксида титана каталитическое количество. Тогда метан уйдет в первом цикле, а откуда брать материал для второго? Да, так и есть, в этих случаях алкоксида титана и метильного гриньяра берут стехиометрическое количество, а иногда даже избыток. Но в сложных синтезах именно органические реагенты, из которых строится цель синтеза, представляют ценность, а титан жалеть никто не будет. Такая вот экономия атомов получается на практике.
Ещё раз про подмену олефинов в Кулинковиче - в чём фишка
Почему реакция замещения в координационной фрагмента Су на олефин идет хорошо? (я бы ответила, что высвобождающийся циклогексен термодинамически стабильнее, вследствие того, что это дизамещенный алкен, но тогда я отвечаю себе же – а если вводим дизамещенный алкен?)
Здесь дело не в том, что реакция обмена олефинов (olefin swap) идёт хорошо, а в том, что, во-первых, эта реакция обратима, и во-вторых, реакция титанациклопропана, образованного дизамещенным олефином, со сложным эфиром идёт плохо, с очень низкой скоростью. В классической реакции Кулинковича со сложными эфирами с самого начала использовались только терминальные олефины (и соответствующие гриньяры), потому что реакция с карбонильной группой сложного эфира очень чувствительна к стерике. Мы, простоты ради, записываем эту реакцию такого какбыгриньяра с карбонильной группой, но понятно, что в химии преходных металлов реакции происходят в координационной сфере металла. То есть всё равно сначала карбонильная компонента должна связаться титаном в комплекс, скорее всего дигапто-типа, и уж в этом комплексе и происходит внутрисферное перемещение электронной плотности, что мы и воспринимаем как реакцию нуклеофильного присоединения к карбонильной группе.Вот здесь первая фишка – если в титанациклопропане есть заместители, стерические взаимодействия делаю связывание сложного эфира невыгодным – равновесие сдвинуто влево. Сама реакция внутрисферного нуклеофильного присоединения необратима, но до неё ещё надо добраться.
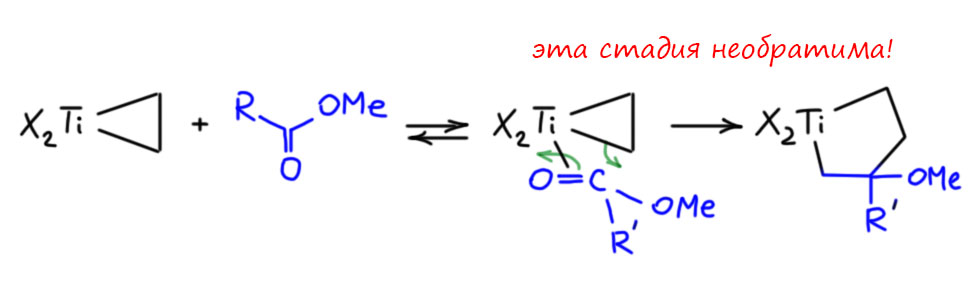
Теперь представим себе, что в реакционной смеси есть олефин с более доступной двойной связью – монозамещенный терминальный олефин (с другими это просто не работает в сложноэфирной версии реакции Кулинковича). Тогда у нас есть система равновесий, и одна быстрая реакция, уводящая новый титанациклопропан в продукт реакции. Это весьма типичная ситуация в химии – равновесие, и необратимая реакция, но только на одном из концов равновесия. Туда и уходит реакция, это такой канал, который забирает всё. Работает весьма надёжно, потому что в этом суть равновесия, даже неважно, какова константа равновесия, велика она или невелика. Хоть единица, хзоть нечто меньше единицы, но не на порядки.

Циклогексен или циклопентен в титанациклопропане вполне эффективны в том, чтобы не давать приблизиться карбонилу сложного эфира. Это только кажется, что это такие маленькие, компактненькие молекулы. Ничего подобного – метиленовые группы по обе стороны от двойной связи, сохраняющиеся в металлациклопропане, своими атомами водорода сверху и снизу оказывают немаленькое стерическое отталкивание, не дают карбонилу связаться в хороший комплекс. Кислород может цепляться за весьма кислотный атом титана, но углерод с заместиелями остается снаружи – то есть там скорее обычный моногаптный комплекс по кислороду, чем дигаптокомплекс, в котором карбонил активрован к реакции.
Более того, чтобы эта методика работала, обычно используют немаленький избыток циклоалкильного гриньяра, и ни о какой каталитической реакции речь вообще не идет – и титан, и вспомогательный гриньяр в избытке относительно олефина и сложного эфира. И даже при этом никакого побочного циклопропанола из вспомогательного гриньяра не образуется.
Так что же такое всё-таки каскад? И еще раз про типы многостадийных процессов
Не только у Вас. Это не самая простая вещь, к тому же малость двусмысленная. И по этому поводу нет никакого консенсуса в литературе и среди ведущих исследователей в этой области. Есть несколько типов многостадийных процессов и с участием комплексов переходных металлов, да и вообще в органической химии. Я предпочитаю очень чётко разделять три типа – one-pot, тандем, каскад. И я не одинок и не сам это придумал, скорее просто систематизировал употребление этих терминов.
Итак, ещё раз. One-pot – это самое простое. Это просто когда две или больше реакций делают в одном горшке. Это как обедать в полевых условиях, когда не хочется мыть посуду, и в одну плошку кладут друг за другом все смены блюд, включая тирамису, и всё это употребляется одной ложкой, которую можно между сменами блюд сполоснуть в луже и протереть листом лопуха. Вот так и тут. Для нас хрестоматийный пример, лучше которого не придумаешь, это последовательность Мияуры и Судзуки-Мияуры. Мы ждём, когда закончится борилирование, и тогда добавляем реагент, который запускает обычное кросс-сочетание – более сильное основание типа карбоната калия. Этот пример вообще очень хорош, потому что тут почти все компоненты реакционной смеси остаются на вторую смену, но вторая реакция всё равно не начинается, пока не добавят еще один реагент. Это значит, что реакции разведены во времени, это разные реакции, мы могли бы не добавлять карбонат калия и тогда остановились бы на первой; мы могли бы выделить бороновую кислоту и зарядить кросс в новом чистом горшке. В этом фишка – реакции-то, в принципе, независимы, а нам просто не хочется мыть посуду и листа лопуха, как назло, рядом нет. У вас растут в лаборатории лопухи? У меня скоро начнут, но тоже пока нет.
Тандем, на первый взгляд, очень похож. Я сам иногда забывают, что есть что, и начинаю заново думать, не баловство ли это со словами. Но нет, всё в порядке. Тандем – это тоже отдельные реакции в одном горшке, но здесь они сами решают, когда начинается следующая. Это значит, что в смеси с самого начала присутствуют все компоненты. С обедом на природе тут аналогия вышла бы скверная, потому что тирамису в супе, даже типа веллютата из спаржи, это как-то слишком смело. Это, пожалуй, лопухом пришлось бы заедать, иначе мог бы случиться конфуз. У нас на сайте, в лекции про карбонилирование в разделе про гидроформилирование, есть хороший пример – гидроаминометилирование. Все компоненты – олефин, амин, родиевый катализатор, синтез-газ – положили сразу, но процесс идёт так, что сначала идёт родий-катализируемое гидроформилирование, образующийся альдегид даёт с амином енамин, который и гидрируется родиевым катализатором. Видите разницу – все эти реакции идут с разными скоростями, но каждая молекула с предыдущей стадии тут же включается в следующую, хотя перевая реакция еще идет и производит новые молекулы альдегида. Создать тандемный процесс намного сложнее, чем просто одногоршковый, потому что вы должны продумать дело так, чтобы продукты следующих стадий не мешали предыдущим, не отравляли бы катализатор, чтобы реагенты были совместимы и не портили бы друг друга, и чтобы можно было в достаточно широком диапазоне варьировать реагенты: в нашем примере брать разные олефины и амины. Тандемные процессы не так распространены, хотя и встречаются достаточно часто. Как правило, хороший тандемный процесс становится новым методом синтеза, особенно если у него достаточно широкий синтетический диапазон. Отдельные реакции в тандемном процессе могут быть и каталитическими и некаталитическими, использовать разные катализаторы. Кстати, в нашем примере, первая и третья стадия используют комплексы родия, но никто нам не сказал, что одни и те же. Мы же уже научились не путать предкатализатор с катализатором, не правда ли. Ещё один признак тандема – каждая стадия всё равно самостоятельна. Мы могли бы прервать процесс на любой стадии, не положив один из реагентов (например, в упомянутом процессе не положили бы амин, остановились бы на гидроформилировании). И мы могли бы начать процесс с любой стадии (например, вместо олефина взяли бы альдегид). И каждая стадия оставляет за собой след – обычно можно брать пробы при недостаточном времени реакции и видеть промежуточные продукты в разных соотношениях.
Каскад – самый интересный вид многостадийного процесса. Это почти всегда каталитический процесс, особенно когда речь идёт о комплексах переходных металлов. А что, бывают каскады без этого? Ещё как, в обычной органической химии пруд пруди каскадов и среди кислотно-катализируемых реакций, и среди свободнорадикальных процессов, и среди всяких согласованных реакций. Например, разные длинные полиены отлично претерпевают каскадные циклизации под действием кислотных катализаторов. Это даже происходит не только в лаборатории, но и в Природе – биосинтез терпенов и стероидов часто использует такие процессы, так что это даже с полным правом можно назвать биомиметической химией. Протонирование одной из двойных связей, даёт карбокатион, который присоединяется к второй двойной связи, даёт следующий катион, он присоединяется к третьей, и последний карбокатион атакует атом кислорода. Вуаля – одним протоном хоть и не семерых (семерых тоже можно было, если бы полиен был подлиннее, и такие примеры есть, мне только лень это вырисовывать), то троих. Даже четверых – три двойных связи и кислород.
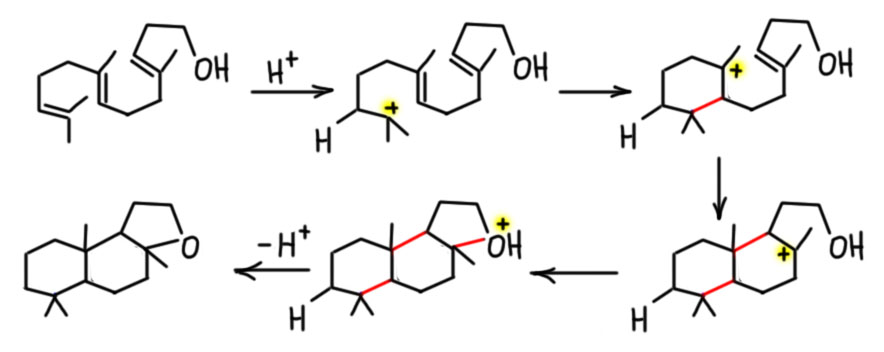
Хорошо видны признаки каскада – в начале возникает реакционный центр (здесь карбокатион), и дальше происходит несколько переносов реакционного центра, пока не исчерпаются доступные места для атаки. Это такой процесс, где каждый следующий шаг цепляется за предыдущий. Может ли быть неполное превращение? Да, безусловно, и не может быть сомнения, что из реакционной смеси можно выделить продукты такого неполного процесса, но это, как несложно понять, будут не карбокатионы, а продукты или элиминирования, или рекомбинации с растворителем. А вот можно ли будет от этих продуктов вновь возобновить каскад это очень даже не гарантировано. Скорее всего, нет. В любом случае это не будет очевидно. Это очень важный признак каскада – неполный каскад может быть легко просто потому что каждая следующая стадия имеет свою константу скорости и свою вероятность. Но прервать каскад и вновь его инициировать или невозможно, или очень трудно.
Каскад всегда состоит из инициирующей стадии – здесь протонирования двойной связи. Цепочки продолжений (переносов реакционного центра) – здесь это алкилирование двойной связи карбокатионом. И завершающей (терминальной) стадии – здесь это алкилирование спиртовой группы карбокатионом.
В химии с переходными металлами роль реакционного центра всегда играет переходный металл. Здесь есть уже сложность в сравнении с обычной химией. Мы и в обычном каталитическом цикле всегда видим непростые приключения этого центра. Кроме того, нас научили смотреть на реакцию именно с точки зрения переходного металла, а не органики, поэтому нам трудно точно понять, что значит перемещение реакционного центра – раз мы считаем, что металл это центр всего, то это все остальное как-то движется относительно этого центра, а не этот центр движется. Это действительно сильно осложняет анализ ситуации. Поэтому попробуем посмотреть на ситуацию чуть-чуть по-другому. Возьмем достаточно простой процесс, который мы называем каскадом – карбонилирующее кросс-сочетание.
Во-первых, мы берем все необходимое сразу и кладем в горшок, один горшок. В этом смысле это похоже на тандем. Но это не тандем. Почему?
Потому что это начинается как реакция кросс-сочетания, могло бы завершиться как полноценный цикл кросс-сочетания. Но не завершается. Вместо этого каталитический цикл продолжается совсем другой реакцией – карбонилировоания. Получается такой общий и единый цикл, который составлен из кусков двух других циклов. Можно ли его прервать на полдороге. Можно – будет просто кросс-сочетание. А можно с середины начать? Нет, потому что там промежуточный комплекс, а не полноценный полупродукт. Как я в этот комплекс попаду, чтобы с него начать? Нет, этот процесс нельзя разобрать на полноценные реакции. Поэтому это не тандем. А что это тогда?
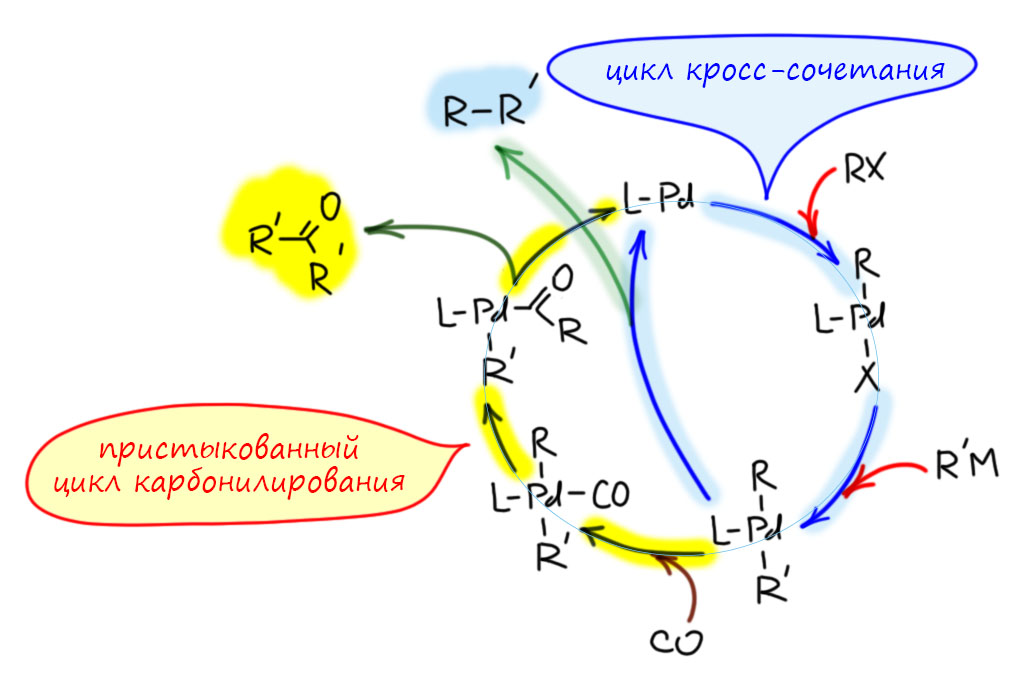
Есть два варианта ответа. Ответ первый – это просто новая реакция, новый каталитический цикл. Это правильный ответ, и так вполне можно думать и даже говорить это вслух и писать в статьях. Это совершенно легитимный способ рассматривать реакции в химии переходных металлов. Проблема этого способа в том, что таких реакций скоро станет чрезвычайно много, потому что соединять можно много чего. Поэтому есть и другой подход – называть это каскадным, составным процессом. Это дает тот выигрыш, что мы не множим процессы, а разбираемся получше в устройстве более простых реакций и потом учимся их соединять, комбинировать, каскадировать. Освоим профессию монтажника каталитических циклов. Это довольно редкая профессия.
Смотрим на этот процесс с этой стороны. И отмечаем две вещи. Первая – перенос реакционного центра. Здесь это не так очевидно, но всё же читается без труда. Было кросс-сочетание. Реакционным центром был σ-комплекс палладия с какими-то органическими лигандами (арилами, винилами, алкилами). И там должен был и остаться до стадии восстановительного элиминирования. Но снаружи пришла окись углерода, и реакционный центр переезжает на карбонил, затем на ацил – на тот фрагмент, которого не было в исходном каталитическом цикле. И только тогда ннаступает развязка. Что мы имеем? Составной цикл (каскад) из двух частей – инициирующей (кросс-сочетание без завершения) и завершающей (карбонилирования, но без обычного начала, вместо обычного начала – незавершенный цикл кросс-сочетания).
Это типичный простой каскад из двух частей. Таких каскадов очень много. Соединять можно многие вещи, можете сами попробовать придумать новую реакцию.
Но если бы на этом дело заканчивалось, игра бы не стоила свеч. Можно было бы и правда просто включить все эти более сложные реакции просто как новые каталитические циклы.
Фокус в том, что последовательность инициирование-завершение можно расцепить в середине, и вставить туда ещё одну реакцию, а значит, как учат нас в курсе матанализа Больцано и Коши, и бесконечное множество реакций. Ну не бесконечное, но достаточно большое.
У промежуточных стадий должно быть одно свойство – каталитический активный центр не должен гибнуть, возвращаясь в начало цикла. То есть для промежуточных стадий годятся не все элементарные реакции. Лучше всего годятся реакции миграционного внедрения, хотя это и не обязательное требование. Именно поэтому множество каскадов основано на вставке в середину цикла стадий карбопалладирования или карбонилирования. И ещё одно пожелание (не требование!) – лучше всего, если все эти вещи будут в одной молекуле. Попробуем для начала сделать так, чтобы они были в разных молекулах. Вставим карбопалладирование в уже готовый каскад кросс-сочетание-карбонилирование. 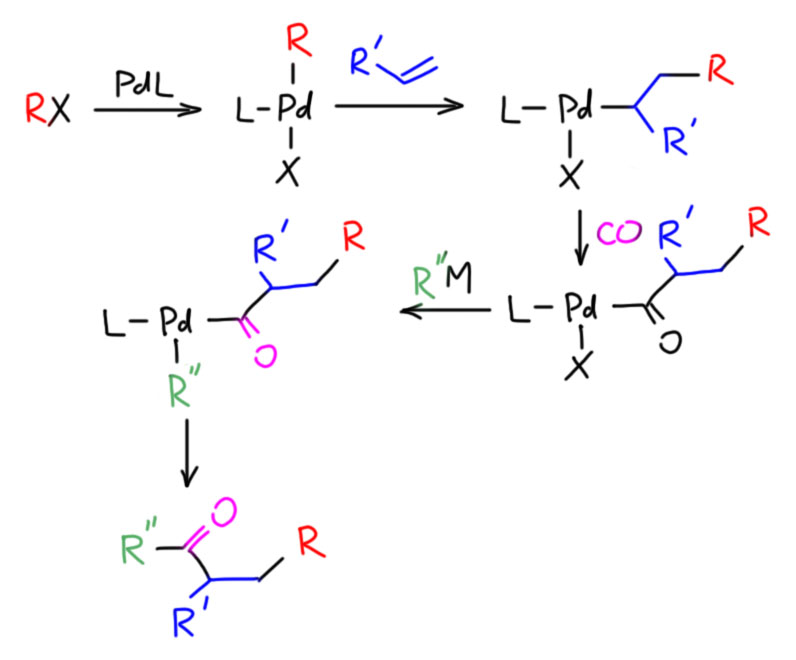
Получается с виду красиво. Но – а кто нам сказал, что монтаж пойдёт именно так. Что переметаллирование не встрянет перед карбопалладированием. Что на любой из стадий все не закончится тем же Хеком, или карбонилирующим Хеком. Здесь вариантов как грязи. Все реагенты изначально присуствуют в смеси и палладий волен выхватывать их в любом порядке, и выбрасывать волен любые.
Поэтому таких межмолекулярных каскадов очень мало, а если они и есть, они очень короткие. Гораздо лучше, когда хотя бы часть событий происходит в одной молекуле – внутримолекулярно, и так. чтобы в результате образовывались циклы, желательно любимые органикой 5 или 6. Ну вот сконструируем каскад из карбонилирования, с передачей реакционного центра в карбопалладировании и с завершением в кросс-сочетании, но так, чтобы основные стадии произошли внутримолекулярно.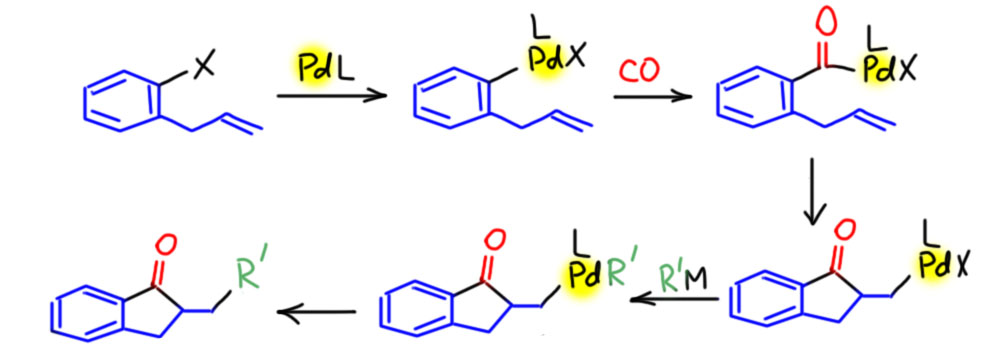
Понятно, что и тут много возможностей для неожиданнго развития и преждевременного завершения. Это вообще непросто планировать. Поэтому за каскады берутся немногие мастера этого дела, имеющие большой опыт и наработанные годами представления о том, как идут такие процессы. И прежде чем что-то делать, моделируют путь реакции расчетами, как минимум чтобы видить геометрию молекул и интермедиатов, как максимум в последние времена вообще обсчитывая разные пути реакций и выбирая оптимальный.
Итак, мы пришли к выводу, что каскад это многостадийный процесс, в котором, как в тандеме, все реагенты изначально присутствуют в реакционной смеси, но описываемый единым каталитическим циклом, с одным каталитически активным комплексом, всегда состоящим из
- стадии инициирования, для чего годится почти любая реакция из нашего множества реакций, обеспечивающих вход в координационную сферу. Очень часто это окислительное присоединение, но может быть и металлирование, в том числе направленное;
- необязательно – стадий передачи реакционного центра (металла с лигандами). На эту роль особенно хорошо подходит миграционное внедрение – карбонилирование или карбометаллирование. Общая характеристика таких стадий – реакционный центр (металл с лигандами) остается в игре, пока не исчерпаются возможности вступать в такие реакции
- стадии терминирования, последней реакции, завершаюшейся восстановительным элиминированием, протолизом, расщеплением ацильного комплекса, бета-элиминировоанием. Образуется продукт и каталитически активная частица регенерируется для нового цикла.
В литературе ещё чрезвычайно популярен термин домино-реакция или домино-процесс. Как правило, это просто синоним каскада, но бывает, что и тандемы называют этим словом. Строго говоря, это не термин, а эмоциональный лейбл – автор процесса так выражает восхищение тем, как классно у него идет процесс и как быстро и надежно чередуются стадии. Источник этого термина прост – домино-эффект, как называют популярную забаву выстроить друг за другом стоя костяшки домино, и уронив самую заднюю, наслаждаться зрелищем, как все они валятся. В сети полно видосов про это, кто-то не поленился выстроить 20000 костяшек, правда получил такое количество просмотров в ютубе, что явно внакладе не остался. Изобретатели термина поэтому никак не характеризуют природу процесса, но просто обращают внимание на общую эффективность. Поэтому я не рекомендую пользоваться этим словом, но привыкнуть классифицировать как одно из трёх one-pot – тандем – каскад. Есть и еще более аккуратная классификация, но она не прижилась, и никто её не использует.
Ещё раз про стереохимию окислительного присоединения
Да, в той первой статье Васка обнаружил саму реакцию окислительного присоединения водорода и галогеноводородов, но не определил стереохимию продукта. В этот момент ещё методов не было, который позволили бы это сделать. Через 4 года он же смог найти метод – это были всякие тонкие эффекты в ИК-спектрах. Сейчас такими сложными и косвенными методами уже мало кто пользуется, но в тот период химики не могли ждать, пока появятся новые спектроскопии и станет эффективным РСА, и очень здорово насобачились извлекать максимум информации из ИК-спектров, а этот тип спектроскопии фактически был первым, он появился ещё до войны, и настоящие знатоки ИК-спектров извлекали массу информации, анализируя эти ужасающие картинки, где полоса сидит на полосе и полосой погоняет. Васка определил, что образуется комплекс с цис-расположением гидридов и транс-расположением фосфинов (Vaska, L. J. J. Am. Chem. Sос. 1966, 88, 4100).
Надо заметить, что при окислительном присоединении любой молекулы типа A-B, новые лиганды А и B оказываются в цис-расположении в координационной сфере металла. Но комплекс, образующийся сначала, не обязательно останется в такой конфигурации. Мы отлично знаем это по рекциям окислительного присоединения галогенпроизводных, с которого начинаются каталитические циклы кросс-сочетания, карбонилирования, Хека и т.д. Но транс-аддукты всегда образуются при изомеризации первончально образовавшихся цис-аддуктов. Механизмы таких изомеризаций бывают разными, обычно это просто вариации на тему “замещение лиганда”, ассоциативно-диссоциативные или диссоциативно-ассоциативныею Нам это не так важно, потому что это не требует каких-то особенных размышлений и редко серьёзно влияет на саму реакцию.
Аддукт с водородом квадратного комплекса Васки конфигурационно устойчив, выделяется и сохраняет первоначальную конфигурацию. В третьем ряду переходных металлов вообще комплексы обычно менее лабильны. Мы уже говорили, что для катализа это не всегда хорошо – слишком они ленивы. Поэтому впервые окислительно присоединение обнаружил Васка на комплексе иридия, но быстрое гомогенное гидрирование открыл в те же годы Уилкинсон на очень похожих комплексах родия из той же группы, но рядом выше.
Присоединение водорода к комплексу Васки очень легко понять, если представить себе превращение квадрата в октаэдр. Совершенно очевидно, что сначала образуется пирамидальный η2-комплекс, после чего back-donation разрушает связь между атомами водорода, и один из гидридов оттесняет самый слабый лиганд хлорид в аксиальное положение. 
Второй вопрос – почему не останавливается на дигапто-комплексе, а идёт окислительное присоединение – предполагает обычный в химии ответ. Потому что такова реакционная способность и энергетика. Дигапто-комплексов водорода, которые в координационной химии обычно называют неклассическими гидридами, потому что когда эта химия начиналась, никто и предположить не мог, что молекула водорода целиком может оказаться лигандом, с тех пор наполучали огромное количество с самыми разными переходными металлами. Иногда они превращаются в обычные дигидридные комплексы, иногда нет. Кроме окислительно присоединения, к таким комплексам применимо внутрисферное депротонирование – молекула водорода в виде лиганда тоже оказывается более сильной кислотой Бренстеда-Лоури, как мы это неоднократно видели с другими лигандами. Так или иначе роль играют в основном разные факторы – энергетика комплексов (что выгоднее, дигапто или два гидрида), наличие свободных мест в координационной сфере металла или лабильных линадов, способность металла к окислительному присоединению (есть ли электроны для back-donation, и сила этого эффекта, которая может в значительной степени изменяться под действием анциллярных лигандов). Всё это очень важно для разработки катализаторов для гомогенного гидрирования.
Такие же тенденции будут и для других молекул, способных к такому превращению – из дигапто в аддукт. Понятно, что чем проще восстанавливается молекула, а это определяется тем, насколько электроотрицательны части, на которые она разваливается, тем меньше шансов увидеть устойчивый дигапто-комплекс, и тем проще идёт окислительное присоединение. Поэтому вряд ли есть шанс увидеть дигапто-комплекс молекулярного галогена (я никогда не видел, хотя не удивлюсь, если для каких-то металлов d0 это бывает). Есть и другая проблема – высокая электрофильность галогенов, скорее всего, не понравится другим лигандам в комплексе, и могут произойти реакции, не являющиеся окислительным присоединением, а например, электрофильное замещение какого-то другого лиганда.
А что там с серебром?
У многих этот вопрос тоже возникает. Вообще до начала нового века и несколько лет после в переходных металлах в органическом синтезе все было довольно понятно. Палладий вне конкуренции. Если добавить реакции с участием водорода (гидрирование, гидроформилирование, гидросилилирование и еще несколько) полезны рутений, родий, чуток платины. Метатезис уже играл – добавляем молибден и опять рутений. С ранними перходными металлами – титан и цирконий. Как-то непонятно мельтешат медь с никелем, но никак не понять, они это серьёзно, или так, для галочки. А все остальное – мало кто помнит названия этих металлов, а уж чтобы что-то с ними делать – только от отчаяния, типа, не нашел человек своей химии, не повезло, не догнал(а), вот и валяет дурака.
За последние 15 лет все изменилось драматически. Палладием уже занимаются или старые зубры типа Бачуолда, или по инерции. И все остальные элементы попёрли как бешеные. Кто что-то знал про иридий или золото? Теперь это чемпионы синтеза. Творят чудеса. Рений? – пожалуйста, интереснейшие окисления, ниобий и тантал – классно, карбены, и так далее. Даже третья группа, малость обиженная подозрением в том, что это или не совсем переходные металлы, или некоторые уже чуть не f-элементы, а с них и спрос отдельный – неплохо наращивает темп.
И только серебро потерялось. Невезучий элемент, до драгоценности не дорос, из фотографии бесцеремонно выперли, и теперь в катализе отстаёт просто уже неприлично. Ни одной своей реакции! Иногда им пытаются что-то по аналогии подправлять в медных реакциях, потому что химия похожа, но мало что получается. Вообще, если посмотреть на реакции, то серебро попадается нередко, но – хуже оскорбления нет! – как вспомогательный реагент, в основном для активации комплексов других элементов, чтобы вытащить из чужой координационной сферы галогенид и направить чужую реакцию по пути, требующему свободного места у металла или повышенной электрофильности.
Серебро оказалось между медью и золотом. Это интереснейший кейс на тему, а что происходит в группе переходных металлов – чем отличаются первый, второй и третий элементы. Всегда бывает так, что первый и третий отличаются радикально, и применения у них драматически отличаются, реакции разные. Это очень понятно, это очередное проявление старинного диалектического закона про переход количества в качество. Электронная структура с точки зрения названия оболочек одинакова, но огромное учеличение количества внутренних электронов, заряда ядра, извините за выражение “релятивистские эффекты”, большой размер атомов, позволяющий размещать больше лигандов и более сложные лиганды, большая стабильность комплексов – все это запускает другие химии.
Второй элемент всегда находится между первым и третьим. Очень часто бывает так, что химия первого и второго похожа, но второй элемент исправляет проблемы первого, у второго элемента более стабильны комплексы, их легче исследовать, у него проще структура валентных состояний. Поэтому вторые элементы часто самые полезные по объему химии – сравните титан и цирконий, хром и молибден, ну и всю старую восьмую группу из короткой Таблицы.
Но в 11 группе все оказалось против второго элемента. У меди и золота драматически разная химия, медь в новые времена стала чуть ли не чемпионом кросс-сочетания, по разнообразию обошла палладий. Золото – карбофильная кислота Льюиса, стала обслуживать огромное количество циклизаций на кратные связи. А серебро зависло посредине, оно оказалось хуже меди,и совсем не доехало до золота. Оно даже ему не мешает, поэтому так популярны каталитические системы, в которых у золота главная роль, а у серебра второстепеннная. Вот что значит древняя поговорка “ни рыба, ни мясо”.
Будет ли так всегда? Не уверен. Возможно, серебро начнет выезжать на карбеновой химии, в которой много похожего на химию меди, но надо еще найти изюминку. Посмотрим.
Следующий блок вопросов пока в работе.
Здесь зависли несколько тем, которые требуют более подробного раскрытия, поэтому они пока висят без ответа. Рано или поздно этот материал будет или освещён здесь, или в основной части курса.
2.04.2023 – Четыре из пяти вопросов из этого блока нашли свое отражение в лекциях, поэтому здесь я это разбирать не буду. Остался только катализатор Херманна-Беллера – знаменитое недоразумение в химии катализа переходными металлами, и я пока не решаюсь изложить историю этого курьёзного заблуждения. Так или иначе, оно носит имена двух очень крупных немецких учёных, которые один раз немного заблудились, но кроме этого сделали очень много важных исследований. В общем, эта история сейчас забыта и никому в голову не придёт пользоваться этим бессмысленным соединением. Но я однажды все же расскажу эту историю, она довольно поучительна, да и химия, как и любая серьёзная наука, устроена так, что из ошибок извлекает даже больше пользы, чем от очевидно правильных вещей.
Катализатор Херманна-Беллера. Несостоявшееся открытие из которого выросла новая химия. Даже две.
Ждите, разбираются…
Кулинкович с заменой олефина
Ждите, разбираются…
Гомогенное гидрирование
Ждите, разбираются…
Нельзя ли поподробнее про Посона-Кханда
Ждите, разбираются…
Нуклеофилен ли карбен Фишера?
Ждите, разбираются…
Арсины вместо фосфинов зачем?
Стало интересно, в каких ситуациях мощи фосфора может не хватить, и понадобится привлечение мышьяка
Применение трифениларсина в качестве лиганда – след от того периода развития катализа комплексами переходных металлов, который можно назвать “лигандным тупиком”. К 1990-м (в этой химии эти годы тоде можно назвать “проклятыми” – в начале 1990-х в химии переходных металлов наметился кризис недостатка идей и знаний – но к середине этого десятилетия эта область начинает мощно разгоняться). В кросс-сочетании и родственных реакциях (Хеке, карбонилировании) воцарился трифенилфосфин, но все попытки как-то пойти дальше были не очень убедительны. Бидентатные лиганды в то время находили очень ограниченное применение в каких-то специальных задачах, а перебор заместителей на фенильных кольцах трифенилфосфина ни к чему серьёзному не привёл, может быть, за исключением трис(о-толил)фосфина, который очень хорошо заиграл в Хеке. Это отдельная история, которую мы как-нибудь посмотрим в другом месте. И вот тогда вышла статья Витторио Фарины, итальянского химика, работавшего в основном в индустриальных исследовательских центрах, про необычайно сильный эффект двух лигандов, трифениларсина и три(2-фурил)фосфина в реакции Стилле (V.Farina, B.Krishnan J. Am. Chem. Soc. 1991, 113, 9585-9595). Статья очень хорошо зашла и стала очень популярной. Её с тех процитировали не менее 875 других статей, что для экспериментальной работы близко к рекордным значениям. В принципе, эту работу можно считать первым исследованием, в котором было аккуратно и убедительно показано, что выбор лиганда может быть критическим параметром для драматического улучшения результатов реакций кросс-сочетания. Это особенно важно для промышленного применения таких реакций. Ускорение в 1000 раз, а это именно то, что обнаружил Фарина, для синтеза в масштабах производства (сотен килограмм на загрузку – это обычный объём для тонкого органического синтеза в фармацевтическом производстве, до нескольких тонн на загрузку – это характерно для производства современных агро-препаратов) это означает не только радикальное снижение себестоимости, но и упрощение очистки продукта от следов катализатора.
Фарина обнаружил этот эффект на самой простой реакции Стилле иодбензола с винилстаннаном. Перепробовав много лигандов, он увидел только два годных – тех, что дают высокий выход продукта и высокую скорость реакции. В этом месте может возникнуть вопрос – какого чёрта так плохо работает трифенилфосфин? Ведь реакция Стилле изначально использовала именно этот лиганд и всё было нормально, хоть и нуждалось в улучшении. Ответ простой – Фарина взял неоптимальные условия реакции специально чтобы увидеть разницу. Так часто делают в сравнительных исследованиях и это вполне корректно, хотя и оставляет некоторые вопросы. Но, так или иначе трифурилфосфин дает ускорение на два порядка скорости, а трифениларсин на три. 
Почему эти два лиганда так работают? Фарина попытался ответить на этот вопрос, сделав весьма умный эксперимент по ингибированию катализатора избытком лиганда.
Кросс-сочетание - это гомогенный или гетерогенный катализ?
Еще один с виду банальный вопрос, простой и очевидный ответ на который – конечно, гомогенный – не совсем точно соотвествует действительности.
Сразу скажем, что кросс-сочетание здесь – просто архетип, самый типичный и важный представитель всего обширного множества реакций, катализируемых комплексами переходных металлов, являющихся предметом настоящего курса. Поэтому можно так и переформулировать: Что мы изучаем в этом курсе, гомогенные реакции или гетерогенные?
С самого возникновения этой области исследований основная задача формулировалась вполне явно и недвусмысленно – развитие именно гомогенного катализа, даже точнее, катализа в жидких средах, в растворах, хотя это и довольно очевидно – попробуйте представить себе гомогенный катализ в газовой фазе (то есть, и катализатор тоже должен быть газообразным!) или твердой фазе – в этом случае реакционная смесь должна была бы быть или твердым раствором, или аморфной – но последнее в любом случае двусмысленно, так как нет принципиальной разницы между жидкостью и аморфным телом.
С самого начала гомогенный катализ именно противопоставлялся гетерогенному, который уже был отлично разработан и применялся в огромном количестве промышленных процессов в основном в вариантах: твердый катализатор – реагенты в газовой фазе или растворе. Гетерогенный катализ очень хорошо тем, что катализатор легко отделяется от реакционнй смеси и продуктов реакции, что радикально упрощает выделение продуктов и регенерацию катализатора, снимает проблемы с дороговизной некоторых катализаторов, потому что они не теряются, а время от времени регенерируются без затрат на новые количества саого ценного компонента – металла. Но за полвека интенсивной разрабоки и эксплуатации гетерогенных каталитических систем к ним возникли большие претензии – обычно это не очень высокая селективность, почти полная невозможность разработки стереоселективных вариантов, и, саоме главное, из чего проистекает все остальное – в принципиальной невозможности хорошо разобраться в механизме, а значит и в осмысленной разработке – трудно рационально заниматься тем, что мы не понимаем и понять не можем – весь 20 век исследования поведения молекул на поверхности твердых материалов далеко не ушли из-за отсутствия методов исследования. Если вы когда-нибудь видели статьи по исследованию гетерогенного катализа с попытками осмыслить механизм, не могли не поразиться совершенно наивным картинкам – какие-то шарики куда-то там цепляются, сидят на каких-то лесенках и решёточках, и что-то натужно изображают – настолько это контрастирует с тем, как мы привыкли рисовать и понимать настоящие механизмы реакций. Но ничего другого там и не нарисуешь, настолько поведение молекул и атомов на поверхности отлично от нашего понимания поведения тех же молекул в растворах. Только в нашем веке в этом смысле наметились серьёзные сдвиги.
Поэтому пошел гомогенный катализ. С самого начала это было просто райское ощущение – наконец-то исследователь может нарисовать нормальный механизм по всем правилам структурной химии, разделять многостадийный процесс на элементарные реакции, пытаться изучать их по отдельности, выделять или ловить интермедиаты, измерять кинетику и всё такое – уровень понимания процессов сразу уехал очень далеко, тем более что к этому времени огромные успехи сделали и координационная, и органическая химия. Появилась возможность целенаправленно конструировать катализаторы, и с ними идти на штурм крепостей, которые не сдавались – в первую очередь, энантиоселективных реакций. Я очень хорошо помню, что в 1970-х оптическая чистота продукта (энантиомерный избыток) в 5% считался великим достижением, и на полном серьёзе доказывалось, что большей чистот ы принципиально добиться никогда не получится. А через 10 лет 95% перестали считаться чем-то необычным – и ключом оказались те самые гомогенные катализаторы и правильно подобранные хиральные анциллярные лиганды. В результате всего за несколько десятилетий мы получили мощнейшую новую химию, которую в этом курсе и пытаемся хоть немного освоить.
Но, как всегда бывает, никакое решение не становится абсолютным. Быстро стало понятно, что за новые достижения приходится очень дорого платить – катализаторы оказались безумно дороги и из-за платиновых металлов во многих, и из-за специальных и очень дорогих лигандов, и из-за непривычной для практически применимой химии чрезвычайной тщательности и высоким требованиям к чистоте всего и самой технике выполнения экспериментов и синтезов. Стало очень трудно отделять катализатор от продуктов, а следовательно и регенерировать его, и не только регенерировать, но и банально очищать продукт от примеси соединений тяжелых металлов, а это неприемлемо, если синтезе делается не просто так, а для производства, например, чего-то, используемого в медицине. Проблема оказалась не менее острой, чем старые проблемы недостатка селективности.
И вот тогда возникло желание как-то сшить гомогенный и гетерогенный катализ, желательно сохранив преимущества обоих, возник запрос на гетерогенизацию гомогенного катализа. Задачу стали с самого начала решать иммобилизацией гомогенных катализаторов на твердых подложках (сначала просто полимерах, а потомв ход пошли разнообразные современные материалы, которых тоже становится все больше и больше). Такие системы работали, но без блеска, свойственного настоящим гомогенно-каталитическим реакциям, прежде всего потому, что соединить иммобилизуемость и собственно эффективность лигандов и комплексов оказалось намного труднее ожидаемого.
Дальше возникла химия наночастиц, возникла гипотеза, что наночастицы переходных металлов могут быть особенно эффективны в качесве катализаторов, а за счет модификации поверхностных атомов лигандами можно добиться и желаемой модуляции свойств и селективности. Тут же возник вопрос – а наночастица это отдельная фаза или просто очень большая молекула (кластер) в растворе. То есть, это гомогенный катализ или гетерогенный? Ответа на этот вопрос нет, потому что это не сущностный вопрос, а скорее, философский, а значит – каждый волен на него отвечать, как хочет.
Дальше выяснилось, что наночастицы тоже надо как-то отделять и регенерировать. Возникли иммобилизованные наночастицы, и это уже совсем непонятно что с точки зрения разделения фаз.
Парарллеьно стал развиваться фазо-разделяемый катализ – это когда реакция идет в одной фазе (она гомогенная), но после реакции осуществяется разделение фаз, при котором катализатор оказывается в одной, а продукты в другой. И пока такие системы разрабатывали, выяснилось, что и там все не так просто, и что разделение фаз во многих случаях все же происходит во время реакции, а не после нее.
В наше время все стало совсем запутанно. Возникло даже такое направление, как катализ индивидуальными молекулами на поверхности (single-atom catalysis), соединяющий преимущества понимания механизма реакции с помощью известных каталитических частиц, с обычной гетерогенностью – реакции идут на поверхности, но там уже не глупые шаники, а конкретные комплексы и все остальное, как мы любим – каталитический цикл, вход, выход, и т.д. Это очень свежая область, немного обескураживающая громогласностью задач, изощренностью экспериментальных медодов, и отсутствием ответов на простые вопросы, например, о каталитической эффективности таких систем. Но – интересно, даже захватывающе.
А вот и совсем простой вопрос по поводу гомогенности или гетерогенности. Как мы знаем, классическая реакция Судзуки-Мияуры идет в органическом растворителе, к которому добавляют немного водного раствора щелочи или чего-то типа этого. И этот раствор не смешивается, остается отдельной фазой. Вопрос – а где идет сама реакция кросс-сочетания? Если палладиевые комплексы в органичекой фазе, субстраты там же, а совершенно необходимый для активации переметаллирования гидроксид – в одной. Как-то тут тоже что-то важное происходит гетерогенно, скорее всего в режиме фазового переноса, в котором межфазным переносчиком служат комплексы палладия.
В общем, получается, что однозначного ответа на простой вопрос нет, и это просто проблема неадекватности простого разделения гомогенный-гетерогенный для сложной системы явлений, вовлеченных в течение реакций с участием комплексов переходных металлов в катализе.
Поэтому не будем придираться к словам – и займемся вместо этого изучением сути.
Бесполезна ли чернь, палладиевая чернь
Да, чернь очень часто вываливается в реакциях с участием комплексов палладия. Вываливается – очень точное слово, это действительно часто происходит неожиданно – пару секуд назад был прозрачный раствор, все было хорошо – и вдруг, раз – и всё черное, и еще через несколько минут на дне валяются черные крупинки.
Мы знаем, что нульвалентный палладий не обязательно валится в осадок в виде черни. Он не валится в двух случаях:
- Pd(0) стабилизируется хорошими лигандами, в первую очередь, фосфиновыми, но, возможно, и какими-то другими. Такой палладий может очень долго быть в растворе, теоретически бесконечно, но это только в том случае, если раствор тщательно изолирован от доступа воздуха – кислород медленно, но верно разрушает лиганды и быстро или медленно оставит палладий без защиты.
- Pd(0) участвует в каталитической реакции, причём скорость стадии с участием этой формы палладия больше скорости слипания атомов палладия – это очень важный случай, разобранный в одной из лекций под рубрикой “бесфосфиновый катализ”
И в том, и в другом случае палладий может долго оставаться в растворе, но в конце концов, вывалится – или реакция замедлится, или лиганды разрушатся. Те, кто реально делал реакции кросс-сочетания, очень хорошо это знает – вываливание черни почти всегда происходит рано или поздно и означает либо, что реакция закончилась, субстраты израсходовались, и в этом случае палладий, оставшийся без дела очень часто берется за свои собственные лиганды и уничтожает их, оставаясь без поддержки. Либо, что хже, что реакция идет не очень хорошо, что-то пошло не так, воздуха напустили или растворители грязные или еще что-то, но тогда реакция на этом заканчивается задолго до полного расходования субстратов. Нужно проанализировать ситуацию, найти причины и приянять меры.
Как только чернь вывалилась, реакции останавливаются. Чернь – это беспорядочные сростки более-менее крупных частиц палладия, переросших стадию наночастиц, слипшихся в слабые агрегаты и ставшие слишком тяжелыми для того, чтобы держаться в растворе за счет обычного теплового движения. Иногда чернь выпадает на стенках посуды, особенно когда стекло старое, изношенное, сильно потертое мешалками – частицам палладия есть за что зацепиться. Иногда на мешалках, опять таки, когда они старые, потертые, с окисленной поверхностью.
Может ли чернь участвовать в реакциях кросс-сочетания или похожих. Да, может. Частички черни – это не кристаллы металла с правильной поверхностью, а реально сростки субмикро- и больших наночастиц, там очень сложный и нерегулярный состав, и много мест, где поверхность огромна, атомы палладия торчат во все стороны во всяких дефектах и прочих наноразмерных неровностях. Такой палладий может взаимодействовать с реакционноспособными субстратами, прежде всего иодпроизводными, но и бромпроизводными с акцепторными группами (любимый субстрат всех исследователей, которые хотят доказать, что кросс-сочетание – такая реакция, которую можно провести со всем чем угодно, включая битый кирпич или картофельные очистки – п-бромацетофенон, обладающий совершенно чудовищной реакционной способностью в окислительно присоединении, он способен подхватить миллионную часть палладия и даже меньше, то есть то, что находится н уровне почти неопределимых следов; во всех таких исследованиях оказывается, что реакцию вели в той же колбе или с той же мешалкой. что и нормальную реакцию кросс-сочетания днем ранее). Окислительное присоединение уносит часть палладия в раствор и начинается обычная бесфосфиновая реакция в растворе.
Такими вещами увлекались многие лет 25 тому назад, потом это всем надоело, потому что предсказать результат таких реакций очень сложно – образцы черни, выпадающие в разных экспериментах отличаются друг от друга совершенно случайным образом, и количество случайно доступного палладия может быть совершенно разным в двух параллельных и с виду идентичных опытах. Выходы и время реакции в таких экспериментах непредсказуемы. Участвуют в таких реакциях только избранные субстраты типа того же п-бромацетофенона или иодбензола, и любые попытки распространить реакцию на что-то реально нужное проваливаются. Поэтому эта химия сама собой заглохла практически без следа, хотя иногда бывают курьёзные рецидивы. Это, кстати, поучительно – никогда не занимайтесь сомнительной фигнёй – даже если получится накропать статейку в какой-нибудь невзыскательный журнальчик, проку от нее не будет, и в лучшем случае она будет забыта.