
Кросс-сочетание. Развитие.
Классическое кросс-сочетание основано на использовании нуклеофилов в виде металлоорганических соединений, загружающих нуклеофильный компонент кросс-сочетания реакцией переметаллирования. Это кажется формальным и немного случайным, но на самом деле, это принципиальный признак раннего этапа открытия и первоначального периода этой химии. Поэтому мы называем такие реакции классическим кросс-сочетанием. Это не общеупотребительный термин, и не результат консенсуса в среде исследователей этой химии,но те, кто долго и внимательно следил за развитием этой области, скорее всего, согласятся с такой периодизацией.
Классическое кросс-сочетание основано на реакции переметаллирования для загрузки нуклеофила на металл, и поэтому всегда использует металлоорганические соединения в качестве нуклеофилов, и, за единственным исключением магнийорганики в реакции КТК, это – малореакционноспособная металлоорганика с умеренной или вовсе почти полностью отсутствующей собственной нуклеофильностью и основностью. Мы будим нуклеофильность с помощью переметаллирования – без этого она скрыта. И мы вообще не трогаем основность – она и не проявляется.
Почему это чертовски важно? Потому что собственная реакционная способность (нуклеофильность и основность) не мешаются под ногами, вызывая множество возможных побочных реакций, которые имели бы все шансы стать основными и послать желанное кросс-сочетание куда подальше. Основность мешала бы, вызывая элиминирование, а также образование совсем не тех комплексов переходного металла, которые ожидаются в каталитическом цикле кросс-сочетания. Явная нуклеофильность тоже не подарок – она сильно ограничивала бы круг субстратов, которые можно использовать. Строго говоря, в реакции с магнийорганикой (реакции Кумады-Тамао-Коррью) мы почти всё это видим, и уже выяснили, что эта реакция очень ограничена, но в тех случаях, когда она применима, её спасает огромная скорость кросс-сочетания. А вот с литий-органикой этот номер уже не проходит – кросс-сочетание с литий-органикой не идёт в таких условиях, и требует специальных условий и решений.
Что требуется для того, чтобы выйти за рамки классического кросс-сочетания? Прежде чем начинать отвечать на этот вопрос, зададим другой вопрос – а зачем нужно выходить за эти рамки? Хотя бы потому, что это удобно и сильно расширило бы рамки метода. Но также и потому, что мы договорились, что в современной химии огромную роль играют экономические и экологические аспекты: то, что мы называем “принципом экономии атомов”, ну и всё, связанное с отходами реакций, их токсичностью. Как бы ни было прекрасно кросс-сочетание с олово-органикой, работа с этими веществами очень опасна и требует особой аккуратности. Цинк и бор менее опасны, но ведь эти элементы и всё что на них ещё висит – отходы, и небезобидные, и дорогостоящие.
Гораздо интереснее было бы грузить на металл не металлоорганику переметаллированием, а непосредственно сам нуклеофил лигандным обменом. Но, чтобы это стало возможно, требуется гораздо более серьёзный уровень понимания того, как устроен каталитический цикл, и того, как им можно управлять. Ключевой фактор здесь – анциллярный лиганд. В классическом кросс-сочетании анциллярный лиганд в основном просто держит низковалентное состояние переходного металла. Но уже в ранних работах мы видим единичные, но важные примеры более серьёзной работы анциллярного лиганда – например, контроль побочной реакции гидридного элиминирования. В 1980-е и первой половине 1990-х такие данные стали накапливаться, что понемногу привело к идее о том, что нужно серьёзнее взяться за изучение и целенаправленный дизайн лигандов. Возникло понятие “well-defined catalyst” – то есть не просто некая форма переходного металла, которую мы добавляем в реакционную смесь, а дальше – как будет угодно уполномоченным богам. Что там с ним происходит и как, мы конечно, пытаемся понять и нарисовать, но в чисто гипотетическом смысле. Нет, теперь мы хотим знать, что точно происходит в координационной сфере, закрепив ту её часть, которая не используется лигандами-акторами, красивым, координационно совершенно стабильным лигандом или лигандами (второе – реже, координационная сфера ведь не резиновая). Пошло бурное развитие химии дизайнерских лигандов, почти всегда, на первом этапе, хелатирующих. Оправдался ли этот подход и в какой степени, мы увидим дальше.
Но начнём мы опять в 1970-х, потому что именно тогда появились две первые реакции, в которых нуклеофил вводится не в виде металлоорганики, а как таковой или в виде сопряжённой кислоты. Это очень специфические реакции, ранний успех которых был обусловлен специфическими свойствами этих нуклеофилов – терминальных ацетиленов и производных трёхвалентного фосфора. Эти особые свойства позволили открыть и запустить в синтез важные методы кросс-сочетания, по способу выполнения и особенностям вполне аналогичные классическим методами кросс-сочетания.
Фактически к Большой четверке классических реакций кросс-сочетания (B, Zn, Sn, Mg) примыкает чрезвычайно важный метод кросс-сочетания с терминальными ацетиленами – реакция Соногасиры или Соногасиры-Хагихары, в которой, по крайней мере формально, в качестве нуклеофила используется не металлоорганическое соединение, а непосредственно нуклеофил – ацетиленид-ион, получаемый прямо в реакционной смеси из терминального ацетилена. На самом деле это не совсем так, и этот метод также основан на реакции переметаллирования, но металлоорганика для переметаллирования получается непосредственно в каталитическом цикле.
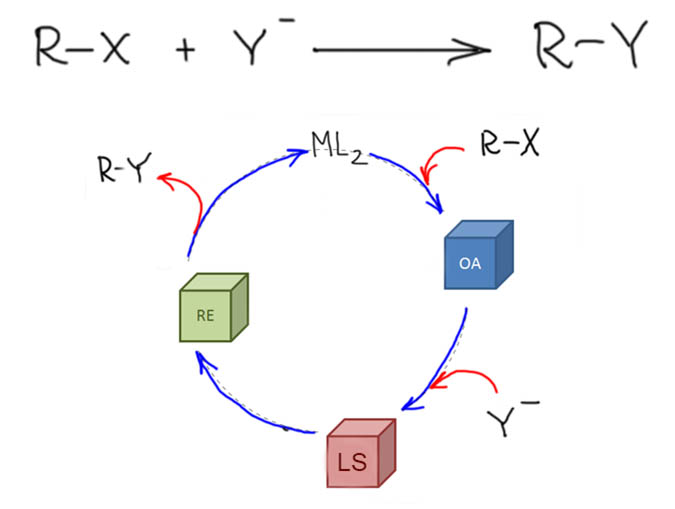
Каталитический цикл в кросс-сочетании с нуклеофилами, не являющимися ковалентно связанными металлоорганическими соединениями, вместо переметаллирования использует реакцию обмена лигандов. В остальном каталитический цикл должен быть похож на обычный, но в реальности в него придётся вносить значительные изменения. На этом слайде показан упрощённый вариант.
Одна из важнейших причин - высокая реакционная способность анионных нуклеофилов, вступающих в множество побочных реакций. Вхождение таких нуклеофилов в координационную сферу металла может происходить не по реакции переметаллирования, а в результате простого замещения (обмена) лигандов, но эта реакция менее управляема, например, возможно вхождение не одной, а двух молекул нуклеофила с блокированием координационной сферы, и т.п. Такие анионные нуклеофилы как карбанионы еще и вызывают множество других побочных реакций, например, элиминирование или некаталитическое замещение.
Задача найти решение этой проблемы, то есть создать катализируемое нуклеофильное замещение, несмотря на явную актуальность долго не решалась.
Терминальные ацетилены - очевидный выбор нуклеофила для кросс-сочетания. Уже на заре органической химии была известна способность ацетиленов образовывать ацетилениды металлов. Особенно легко и практически самопроизвольно образуются ацетилениды меди. Перспективы ацетиленов в каталитической химии также были выявлены очень рано - уже в работах Реппе в 1930-х, и несколько реакций в присутствии комплексов меди вошли в арсенал органического синтеза, показав, что именно медь является вероятным ключом к раскрытию синтетического потенциала терминальных ацетиленов.

В "старой" химии есть довольно много реакций с участием ацетиленидов меди, как предварительно полученных, так и образующихся in situ. Некоторые из них очень похожи на кросс-сочетание, впрочем, не являясь каталитическими процессами.
В реакции Кадьё-Ходкевича происходит кросс-сочетание терминальных ацетиленов с ацетиленовыми бромидами в присутствии соли меди (I) и основания - амина.
В очень похожей реакции Кастро-Стефенса готовый ацетиленид меди реагирует с ароматическим иодпроизводным в присутствии пиридина.
Эти реакции очень хорошо показывают выдающийся потенциал ацетиленовой металлоорганики для создания новых углерод-углеродных связей.
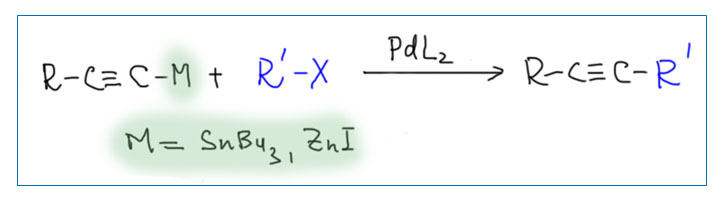
Появившиеся в 1970х реакции кросс-сочетания вполне годятся и для образования связи sp-sp2. Но для этого необходимо заранее или в реакционной смеси получать металлорганические производные ацетиленов.
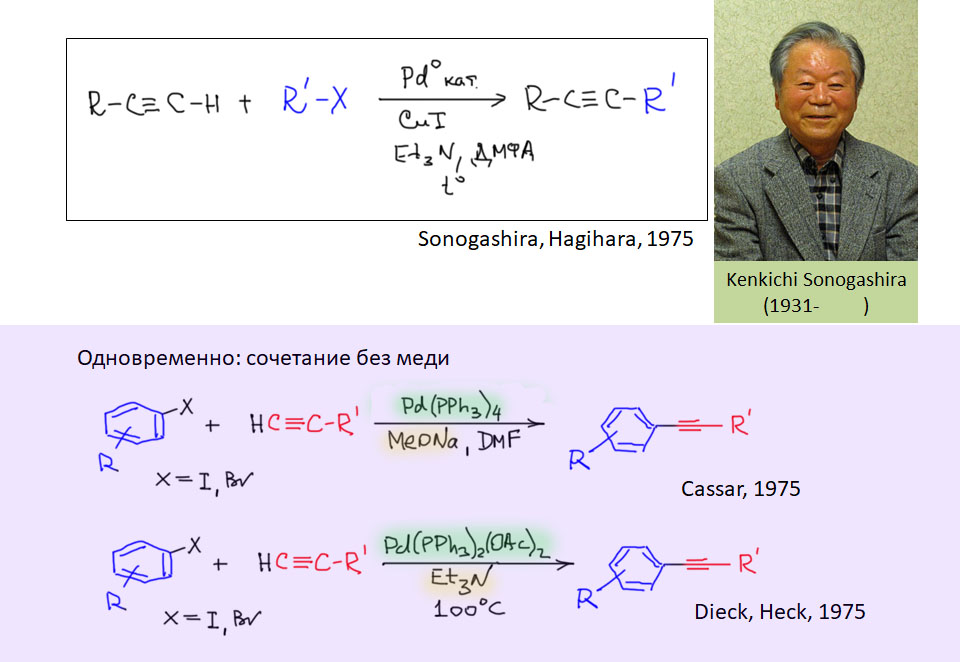
Выдающимся достижением стало одновременное открытие в 1975 году прямой реакции кросс-сочетания терминального ацетилена с ароматическими галогенпроизводными. Луиджи Кассар в Италии и Ричард Хек в США описали реакции в присутствии комплексов палладия и оснований. Японские исследователи в группе Кенкити Соногасиры решили не отказываться от меди и предложили двойную каталитическую систему с комплексами палладия и меди и алифатическими аминами в качестве основания. Именно эта система оказалась наиболее гибкой и универсальной и быстро завоевала популярность, став именной реакцией. При этом, особенно в последние 10-15 лет, когда на первый план стала часто выходить экономичность и простота каталитических систем, стали все чаще и чаще обращаться к "безмедной" Соногасире, как это стали называть (copper-free Sonogashira), которую все же первыми описали Кассар и Хек.
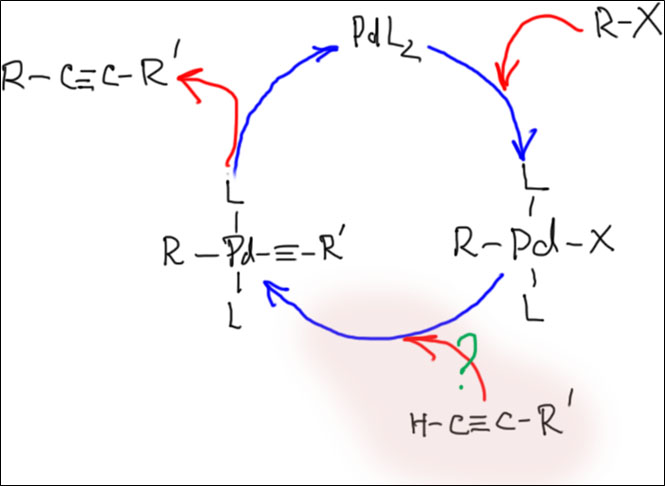
В кросс-сочетании с терминальными ацетиленами, следующем обычному каталитическому циклу с окислительным присоединением в начале и восстановительным элиминированием в конце остается один вопрос - как входит в координационную сферу сам ацетилен и какова в этом роль меди, коль скоро мы закрепляем за палладием роль основного металла, в координационной сфере которого происходит весь спектакль. Почему мы можем использовать достаточно слабое основание (pK алифатических аминов не превышает 12), которое не может депротонировать сам ацетилен (pK на уровне 25)?
Для реакции Соногасиры предложен не один механизм, объясняющий роль меди. Полной ясности нет до сих пор, но наиболее простой и логичной гипотезой является участие в этой реакции ацетиленидов меди. Очевидным путём их образования является депротонирование дигапто-ацетиленовых комплексов меди - координация тройной связи по атому металла приводит к электрофильной активации терминального ацетилена и существенному увеличению CH-кислотности (снижению эффективного pK).
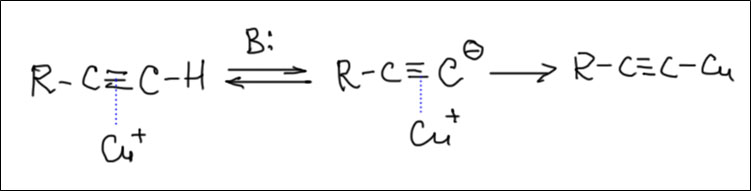
Ацетиленид меди далее участвует в переметаллировании и входит в обычный каталитический цикл классического кросс-сочетания.
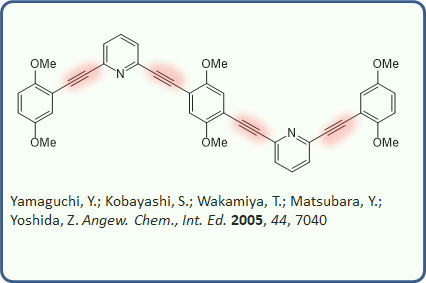
Реакция Соногасиры-Хагихары стала невероятно популярным методом конструирования сложных молекул, особенно таких, которые нацелены на использование в качестве материалов в молекулярной электронике, оптической сенсорике и прочих модных областях, где требуются огромные молекулы, состоящие из функциональных блоков, соединенных в единое целое с помощью фрагментов, обеспечивающих сопряжение между блоками.
Ацетиленовый линкер (linker - связующее звено) совершенно незаменим в этой роли, так как электронная структура этого фрагмента - цилиндрическая симметрия и пара ортогональных пи-связей, не привязанных к конкретной геометрии самого линкера, обеспечивают взаимодействие блоков практически в любой геометрии соединения.
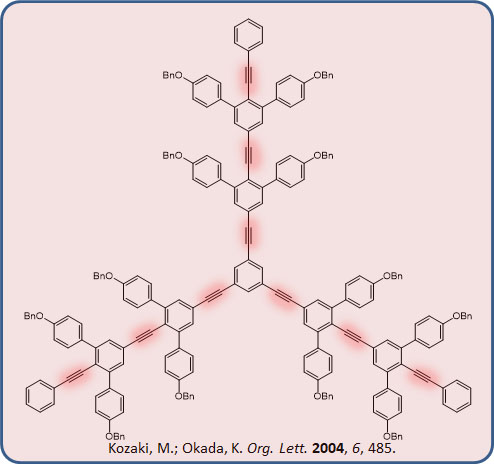
С помощью реакции Соногасиры-Хагихары получено огромное количество таких молекул, и пара примеров показана слайде. Для создания конечной молекулы кросс-сочетание повторяется несколько раз до достижения необходимой архитектуры.
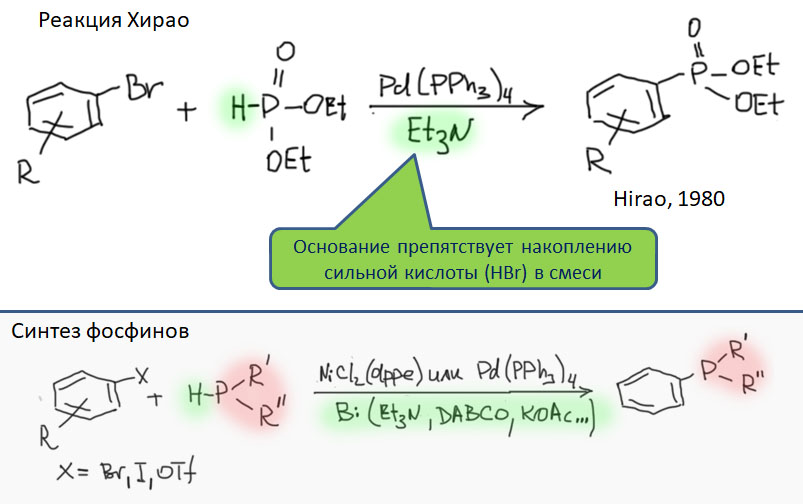
Самая первая реакция, в которой получилось сделать кросс-сочетание с свободным нуклеофилом, если не считать реакцию Соногасиры, в которой все же происходит переметаллирвоание, хотя и скрытое от глаз в каталитическом цикле, - образование связи углерод-фосфор. Ее описал в самом начале 1980-х Тосикадзу Хирао с сотрудниками (Япония). Реакция была быстро расширена на многочисленные нуклеофильные производные фосфора, и позволила получать фосфонаты, фосфинаты, фосфины и другие важные фосфорорганические молекулы.
Каталитические системы для реакции Хирао и близких методов довольно просты: палладиевый катализатор с лигандами первого поколения плюс основание. Роль основания в данном случае не в том, чтобы депротонировать PH-кислоты, так как производные фосфора уже в этом состоянии являются вполне компетентными нуклеофилами и не нуждаются в депротонировании. Тем не менее, после восстановительного элиминирования продукта неизбежно образуется сильная кислота, и ее накопление в реакционной смеси быстро заблокировало бы каталитический процесс. Основание просто подчищает эту кислоту в реакционной смеси (используется термин scavenger - а это буквально и есть "чистильщик, мусорщик", тот, кто подбирает всякую дрянь).
Реакция Хирао и ее варианты - первый метод каталитического кросс-сочетания углерод-гетероатом, реакции, создающей связь углерода и неуглерода, и в этом смысле она продемонстрировала фантастический потенциал этого типа реакции. Дальнейшее развитие методов кросс-сочетания действительно позволило создавать связи углерода с практически любыми неметаллами и металлоидами, а также и с некоторыми металлами.

После открытия основных реакций кросс-сочетания – большой пятерки реакций Кумады, Негиси, Стилле, Судзуки, Соногасиры, а также принципиальной возможности применения кросс-сочетания к созданию не только связи C-C, но и связей углерод-неуглерод случилось весьма длительное затишье, когда найденные реакции применяли, совершенствовали, изучали с большим успехом, но новых прорывов не было. Совершенно очевидной следующей задачей стала разработка кросс-сочетания для создания связей углерод-азот. Потребность в действительно мощном и гибком методе синтеза аминов в синтезе ощущалась чрезвычайно сильно. В это время уже начинает развиваться химия молекул, имеющих какие-либо важные функциональные свойства – полупроводников, сенсоров, люминофоров, сенсибилизаторов и т.д., и т.п. – все то, что получило название химии материалов. В русском языке у этого слова есть устойчивая ассоциация со строительством, и услышав это определение мы невольно начинаем думать о кирпичах и штукатурке, и недоуменно моргать. Но это неуместная аллюзия. В английском material – это самое банальное слово, употребляемое просто для обозначения любой полезной вещи, пусть даже и молекулы. Химия полезных молекул – вот что такое material science. Заглянув в любой журнал с таким названием, мы увидим статьи обо всем на свете, лишь бы это было бы хоть чем-то полезно, имело бы какие-то доказанные свойства.
Так вот, среди полезных молекул, то есть материалов множество производных азота, и это, наверное, неудивительно – сама Природа точно не брезгует этим элементом. Молекул много, в том числе таких, которые можно нарисовать на бумаге, а вот методов синтеза производных азота в органическом синтезе было хоть и много, но с огромными пробелами: множество аминов или вообще недоступны методами классической органической химии, либо доступны, но с такими сложностями, что проще не связываться. Потребность в новой химии была огромной.
Вряд ли, поэтому, можно удивиться тому, что именно C-N кросс-сочетание открывает новую главу в кросс-сочетании и вообще в применении комплексов переходных металлов в органической химии. Открытие этой реакции вызвало совершенно бешеный интерес к применению химии переходных металлов, и буквально за 10 лет после её открытия мы увидели огромные достижения – были решены и многие другие задачи, считавшиеся до этого практически нерешаемыми (например, эффективное вовлечение хлорпроизводных в кросс-сочетание и другие реакции).
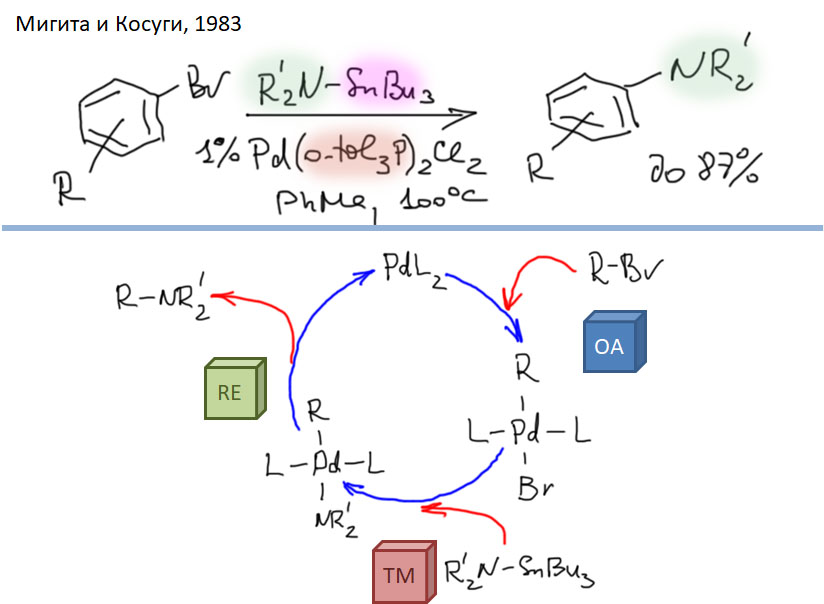
Возможность осуществления кросс-сочетания с образованием связи углерод-азот очень долго не была реализована, хотя и представляет собой важнейшую цель синтеза. Обычная органическая химия умеет синтезировать амины самого разного строения, но далеко не все. До появления методов кросс-сочетания задача введения азотных заместителей в ароматические кольца была практически не решена; известные методы были неэффективны и работали только для узкого числа конкретных примеров.
Проблему пытались решить Мигита и Косуги, использовав полный аналог реакции кросс-сочетания оловоорганики, со-открывателями которого они были вместе с Стилле. Реакция действительно получилась на нескольких простых примерах, но необходимость получения и работы с оловянными производными аминов отбила охоту исследовать этот метод дальше - в синтезе он более почти никогда не встречается. Но в этой работе была сделана очень важная вещь - найден лиганд, который обеспечивает восстановительное элиминирование продукта кросс-сочетания, и этим лигандом оказался хорошо нам уже знакомый трис(орто-толил)фосфин. Стало, по крайней мере, ясно, что нужен стерически объемистый лиганд с большим коническим углом.
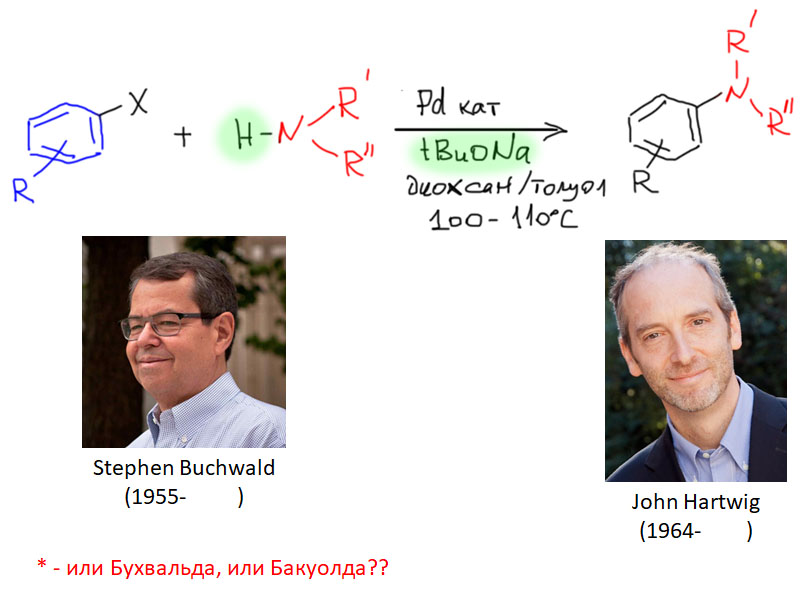
Но проблема оставалась нерешенной еще более 10 лет, пока за нее независимо не взялись две мощные группы в США, Джона Хартвига и Стефена Бачуолда (или Бакуолда, но в России и не только его фамилию принято читать как чисто немецкую - Бухвальд: сам он на это прозвище, по слухам, не откликается). Обе группы стартовали ровно с того места, где дело было оставлено Мигитой и Косуги, но кропотливое исследование механизма и условий привело к неожиданно простому решению - взять сам амин вместо оловянного амида, и обеспечить восстановительное элиминирование за счет отщепления протона сильным основанием. Оптимальным основанием оказался трет-бутилат натрия, а оптимальным растворителем - малополярные толуол или диоксан. В этих средах трет-бутилат натрия плохо растворим, что оказалось очень важно, так как не создается большого избытка сильного основания, препятствующего правильному порядку связывания лигандов в координационную сферу палладия. Первым лигандом остался трис(о-толил)фосфин.
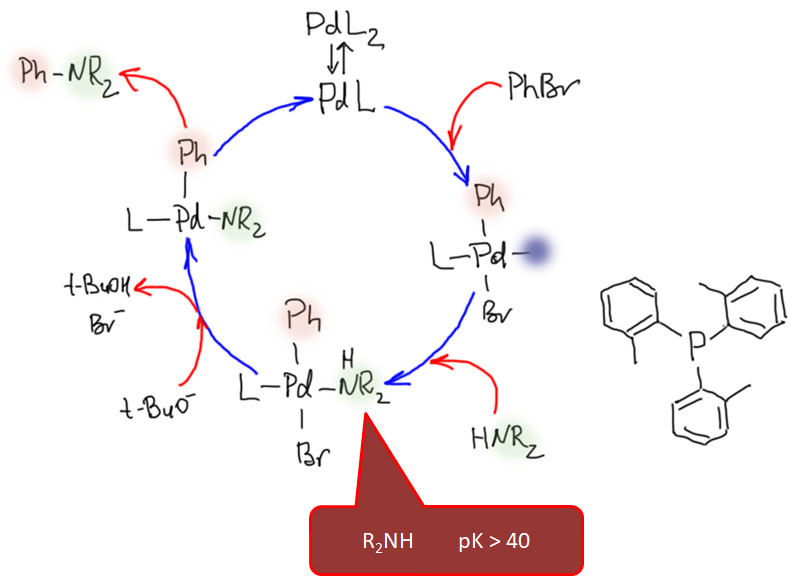
Этот лиганд, который впоследствии обозвали лигандом первого поколения, работал, но не очень хорошо. Кросс -сочетание происходило с достаточно нуклеофильными алифатическими аминами, но когда такой амин входит в координационную сферу палладия, наблюдается весьма противная побочная реакция - гидридное элиминирование, которое приводит к восстановительному дегалогенированию исходного бромпроизводного.
Отметим еще, что депротонирование аминов, являющихся чрезвычайно слабыми кислотами, основанием умеренной силы достигается потому что, когда амин входит как нейтральный лиганд в координацонную сферу палладия, кислотность значительно возрастает за счет активации атомом металла. Похожий эффект мы уже видели в реакции Соногасиры с ацетиленом.
Возникла очень хорошо известная в химии ситуация - есть важная реакция, она вроде бы идёт, и это уже повод для ликования, но как-то не так, как хотелось бы. Без блеска. Выходы небольшие, куча побочных, диапазон ограничен малоинтересными реагентами.
Раньше так бы всё и осталось. Потыкались бы вокруг, да около, да и оставили бы до лучших времён. Но в последние несколько десятилетий развитие химии чрезвычайно ускорилось - появилось много новых методов, накопленные знания вооружили исследователей новыми подходами к анализу данных, да и самих исследователей в мире стало намного больше - в новом тысячелетии химией на весьма серьёзном уровне стали заниматься в полусотне стран.
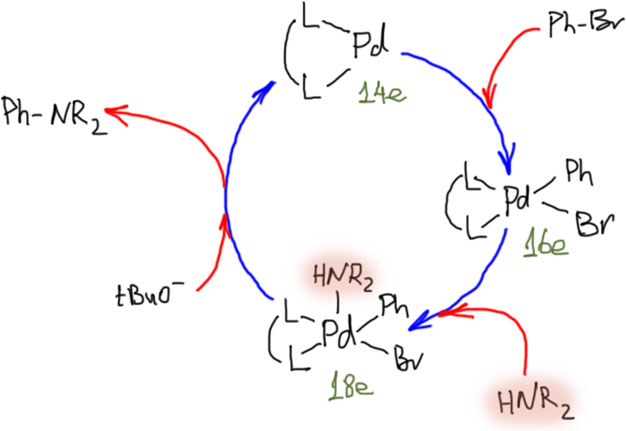
Поэтому первооткрыватели C-N кросс-сочетания (в литературе эту реакцию очень часто и не очень удачно называют аминированием) продолжили штурм без той самой раскачки, о которой мы так много слышали. Решение стали искать в бидентатных фосфинах. Это не очень простое решение, так как если нам нужно заместить галоген у палладия в 16-электронном комплексе без использования переметаллирования, нам придётся расчитывать на ассоциативно-диссоциативный механизм с образованием 18-электронного комплекса. Формально всё хорошо, но для металлов 10 группы такой путь долго считался невыгодным. Но это сработало!
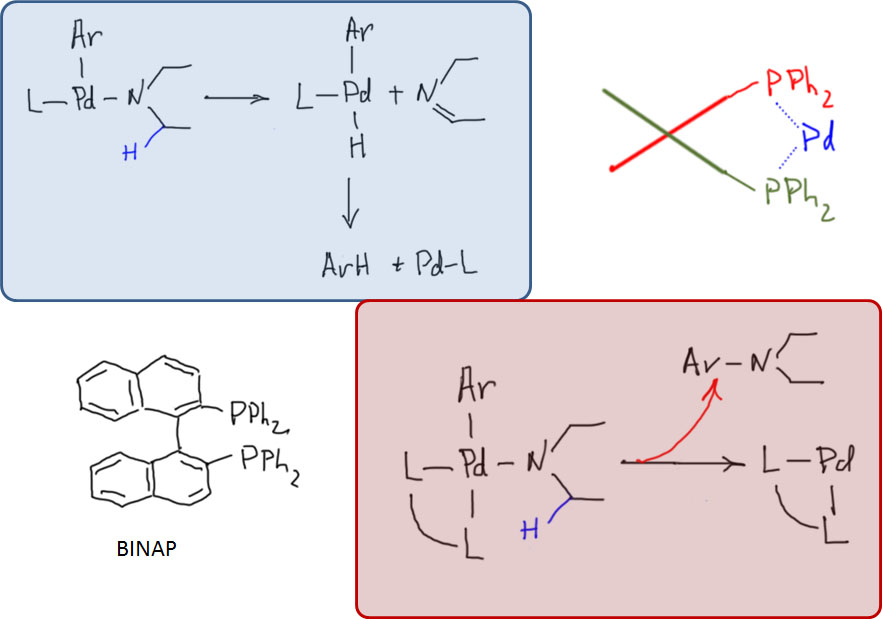
Самая неприятная побочная реакция в этой химии - восстановительное дегалогенирование, происходящее при бета-гидридном элиминировании из амидного комплекса палладия конкурентно с восстановительным элиминированием. Вместо ожидаемого продукта с новой связью C-N образуется банальный продукт дегалогенирования.
Борьба с восстановительным дегалогенированием стала основной целью, и обе группы независимо пришли к одинаковому решению - необходимо надежно заблокировать доступное координационное место у палладия, которое требуется для захвата гидридного лиганда. Если лиганд монодентатный, как фосфины первого поколения, то за счет спонтанной диссоциации в координационной сфере может остаться только один фосфин, а освободившееся место будет использовано палладием для отщепления гидрида. Заблокировать это место можно бидентатным фосфином. Так появились лиганды второго поколения, в качестве которых Хартвиг выдвинул известный нам dppf, а Бачуолд - также известный и уже знаменитый лиганд BINAP. Оба лиганда хорошо работали, но именно BINAP стал основным лигандом C-N кросс-сочетания. И остается им до сих пор.
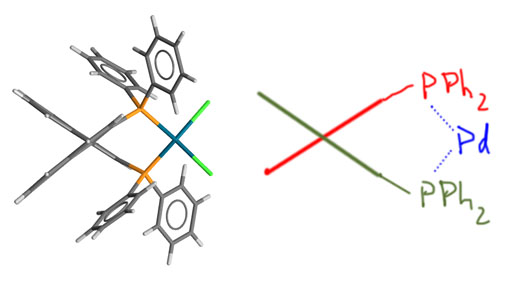
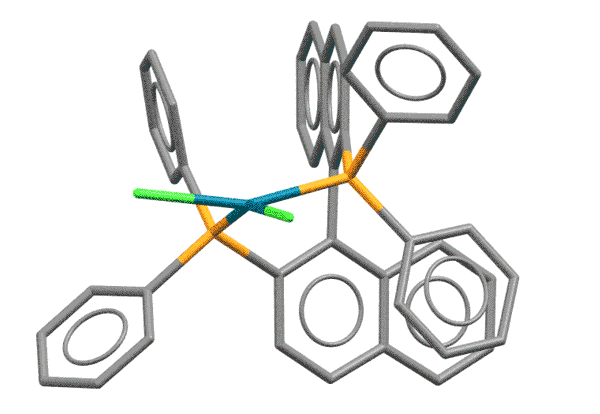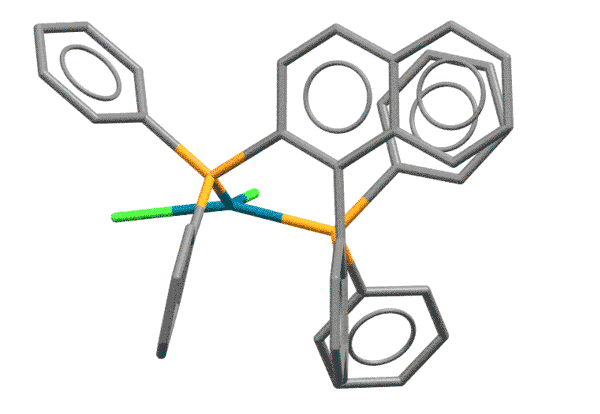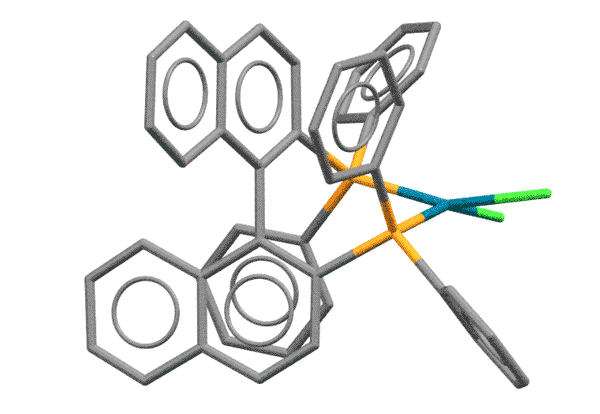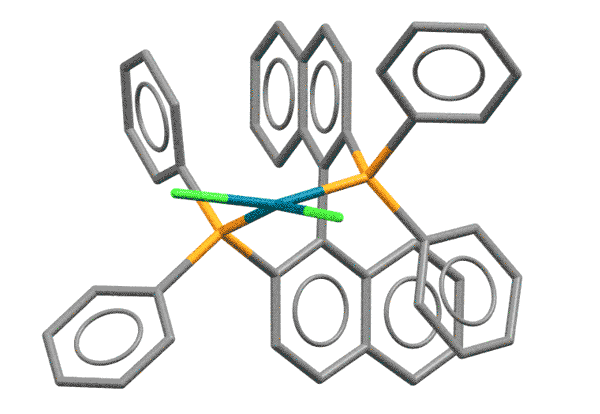
BINAP - это дифосфиновый лиганд на основе 1,1'-динафтила, впервые синтезированный совершенно для другой реакции - энантиоселективного гидрирования. Молекула BINAP хиральна за счет того, что она имеет единственный элемент симметрии, ось второго порядка. Для C-N кросс-сочетания хиральность BINAP не имеет значения, а важно то, что из-за стерического взаимодействия атомов водорода в положениях 8 колец, нафталиновые фрагменты скручены относительно друг друга, как лезвия ножниц. Фосфиновые атомы фосфора поэтому расположены весьма необычным образом, так, что образующийся с палладием хелат оказывается напряженным, с геометрией, значительно отклоняющейся от плоского квадрата. И не забываем про стерический объем лиганда. Комплексы BINAP поэтому отлично работают на выпихивание продукта восстановительного элиминирования, эффективно блокируя координационную сферу металла от гидридного элиминирования.
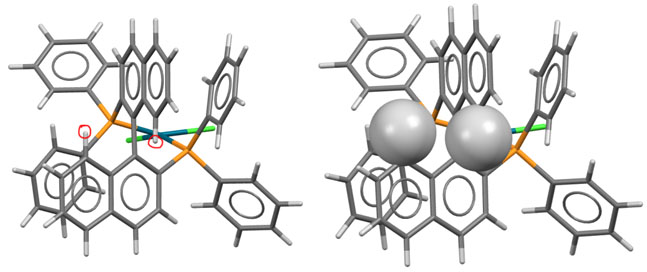
На проекции комплекса BINAP показаны те самые два водорода сферами с радиусами, обозначающими самое близкое возможное сближение. Хорошо видна причина, по которой два нафтила расходятся на такой большой угол. Водород, оказывается, не такая маленькая штука.
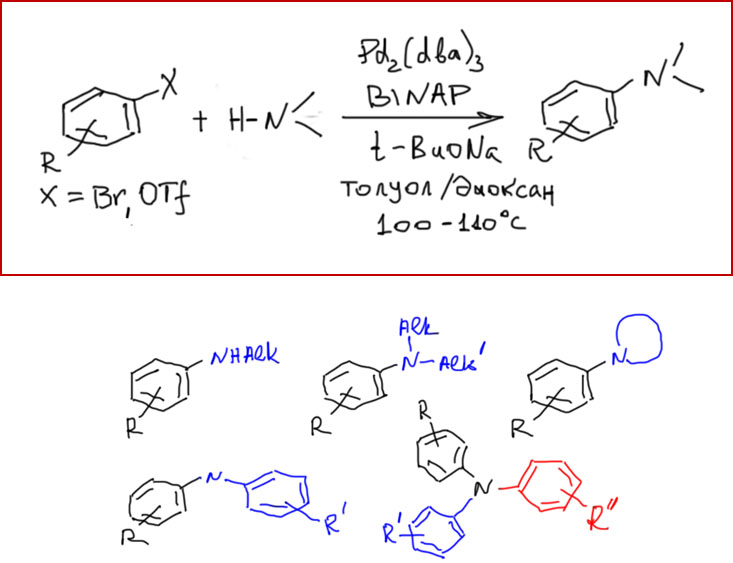
Разработанный метод очень быстро завоевал ошеломляющую популярность в синтезе аминов. Протокол, использующий ацетат палладия или комплекс палладия с dba в присутсвии BINAP, трет-бутилата натрия в толуоле или диоксане при довольно длительном кипячении, стал стандартом в синтезе ароматических аминов, содержащих от одного до трех ароматических колец, а также алкильные заместители в самых разных комбинациях и с самыми разными заместителями в ароматических кольцах. Этот стандарт остается в силе до сих пор, и именно с него обычно начинают поиск условий синтеза новых молекул, содержащих ариламиновые фрагменты.
Но стандартный протокол не всемогущ. Он требует использования ароматических бромидов или трифлатов, но плохо работает для хлоридов или даже иодидов, не говоря уже о более экзотических, а на самом деле самых банальных ароматических электрофилов типа тозилатов, мезилатов и т.п. Протокол требует немаленькие количества катализатора, а это очень дорого. Множество субстратов упорно сопротивляются, и дают плохонькие выходы, и среди них гетероциклические производные, амиды, маленькие амины и сам аммиак. Условия реакции довольно жесткие, что, в частности, не дает возможности использовать соединения, чувствительные к действию сильных оснований, например, енолизуемые кетоны, сложные эфиры, сопряженные кратные связи и т.п.
Возникло острое желание усовершенствования, и за это взялись уже не только Хартвиг с Бачуолдом, но и множество других изобретательных и промысловатых исследователей.

Успехи не заставили себя ждать. Вообще по скорости развития эта реакция, возможно, не имеет себе равных в органической химии - буквально за 10 лет объем информации, количество каталитических систем, лигандов и протоколов, количество реальных синтезов с использованием метода увеличилось на несколько порядков. Этому способствовало и драматическое изменение методики исследований - появился так называемый высокопроизводительный скрининг, когда реакции с множеством варьируемых условий одновременно стали ставить сотнями и тысячами в микроколичествах в автоматическом режиме с автоматическим анализом смесей методами хромато-масс спектроскопии.
Наиболее представительный и широко известный набор лигандов предложил тот же Бачуолд, использовав в качестве основы дифенилы с одним фосфиновым заместителем в положении 2. Совершенно очевидна преемственность этого типа лиганда с BINAP - они активно используют принцип построения знаменитого лиганда, одновременно корректируя его недостатки. В дизайне лигандов для кросс-сочетания используется несколько таких направляющих принципов-гипотез.
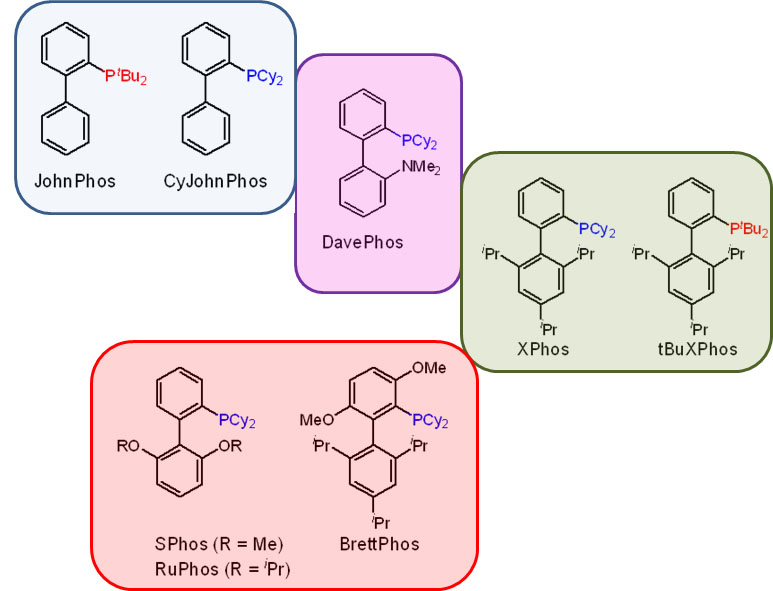
1. Лиганд должен быть сильнодонорным, тогда он эффективнее будет вступать в окислительное присоединение и справится с сложными уходящими группами типа хлора или тозилата. Для этого на фосфоры сажают пару алкилов.
2. Лиганд должен быть объемистым и эффективно выпихивать продукт из координационной сферы. Чем лучше выпихивает, тем больше скорость реакции, и тем меньше нужно катализатора. Для этого алкилы делают объемистыми. В лидерах рейтинга трет-бутилы и циклогексилы.
3. Лиганд должен быть и не монодентатным, и не бидентатным, а чем-то посредине. Как это? Объясню точнее на следующем слайде, но замечу, что для этого лиганд должен иметь затейливую форму, и содержать основной лигандный центр, обычно фосфин, и дополнительное, более слабое место связывания.
Основной ключ к успеху - более или менее целенаправленный синтез и исследование лигандов. Возникло даже такое пафосное понятие - дизайн лигандов. Это, как часто бывает в естественных науках, очевидный перебор. Для настоящего дизайна требуется детальное знание того, как точно работает та или иная реагирующая система. Современные методы исследования, безусловно, достигли огромных успехов. Есть методы, позволяющие в реальном времени следить за составом многокомпонентных смесей; пути и механизмы реакций моделируют с использованием современных и очень быстрых методов квантово-химических расчетов, и т.п. Но химия по прежнему остается слишком сложной для действительно точного моделирования, и успехи здесь только приблизительны.
Обычный путь для "дизайнера" лигандов состоит не в полном моделировании механизмов, а в формулировании гипотезы о том, что сильнее всего влияет на работу каталитической системы, создании на основе этой гипотезы серии пробных лигандов, их тестирование в реальных реакциях и отбор наиболее перспективных лигандов. Это типичный путь проб и ошибок.
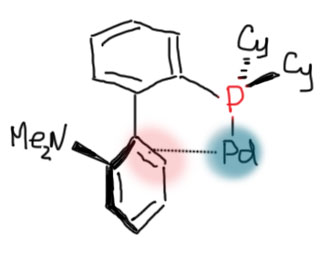
Вот пример того, как устроен комплекс палладия с одним из лигандов дифенильного типа, DavePhos по данным рентгеноструктурного анализа (J. W. Faller, N. Sarantopoulos, Organometallics 2004, 23, 2008-2014). В этом примере на палладии висит еще один лиганд, но это сейчас несущественно. DavePhos сидит на палладии фосфиновым центром, и это очень прочный комплекс, и лиганд в этом смысле является координационно стабильным - он сохраняется пока в координационной сфере развивается собственно каталитическая реакция.
Второе координационное место у палладия связано с вторым кольцом дифенила за счет дигапто-связывания бензольного кольца. Это существенно более слабая связь и во время реакции она может временно разрываться, запуская в координационную сферу другой лиганд, например, амин в C-N кросс-сочетании. Такой лиганд называется гемилабильным, то есть "наполовину" лабильным. Получается почти идеальная ситуация - лиганд эффективно контролирует координационную сферу металла, впуская в нее только полезные лиганды, и блокируя побочные реакции.
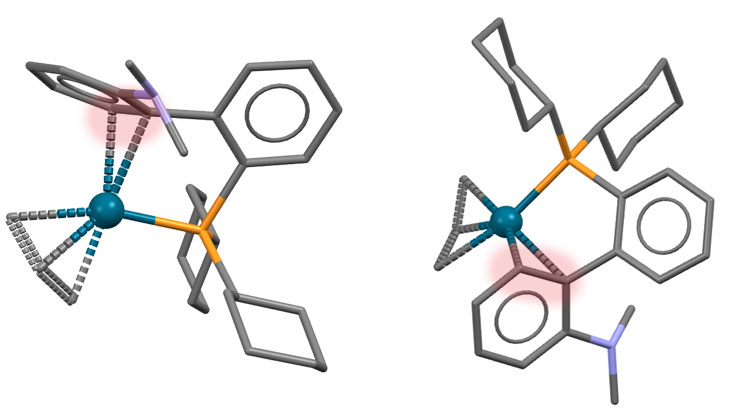
Почему он так поступает ? - Он умный? Это такая молекулярная машина с мозгами или встроенным микроконтроллером и вайфаем - ждет указаний, кого впускать, кого гнать прочь или сама знает? Ничего подобного. В химии роль мозгов и контроллера с вайфаем всегда играют химические равновесия. В данном случае, равновесия лигандного обмена. Стабильный лиганд не обменивается в равновесиях обмена. Лабильный обменивается почти всегда, создавая множество ситуаций в координационной сфере, в частности и побочные реакции тоже. А вот хороший гемилабильный лиганд имеет такие значения констант устойчивости (а обмен лиганда в таких комплексах всегда идет через диссоциацию), которые позволяют ему выбирать, и не обмениваться на все, что попалось, но только на такие лиганды, которые предъявляют конкурентные константы связывания. Важно подобрать лиганд так, чтобы он работал в пользу нужных лигандов и нужной реакции. Чудес, естественно, не бывает, и такие лиганды никогда не бывают универсальными.
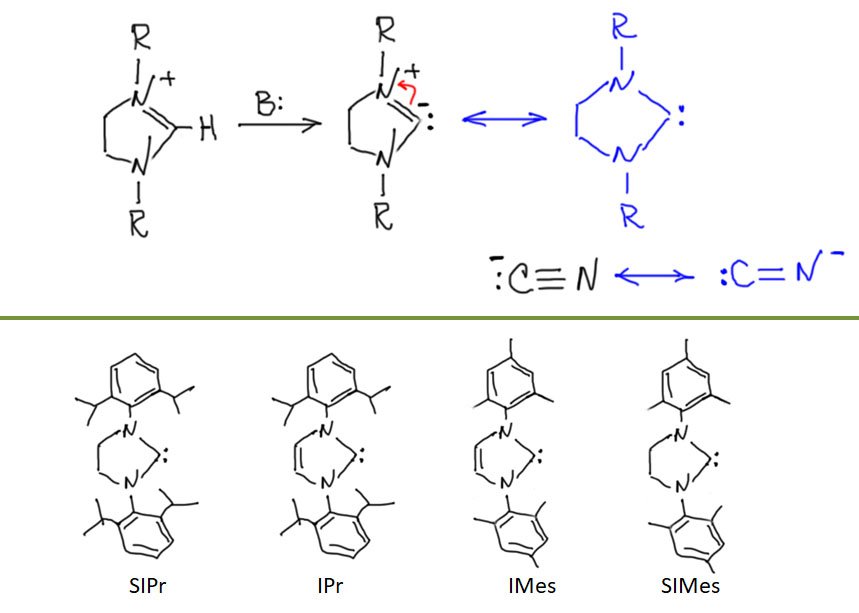
Фосфиновые лиганды практически незаменимы для палладий-катализируемых реакции кросс-сочетания и аналогичных. Но им все же удалось найти некоторую замену, далеко не полную, но весьма впечатляющую по достигаемым результатам. Это класс лигандов, которые называются гетероциклическими карбенами, наиболее популярные из которых получаются изумительно просто - депротонированием легкодоступных четвертичных солей имидазолов. Химически эти соединения являются просто аналогами цианид-иона, имеющего вторую граничную структуру изоцианида с карбеновым характером углерода. Наиболее распространенной в катализе стала уже знаменитая четверка карбеновых лигандов - IPr/SIPr и IMes/SIMes с объемистыми арильными группами. Такие лиганды отлично работают в реакциях С-С и C-N кросс-сочетания, в том числе и с хлорпроизводными.
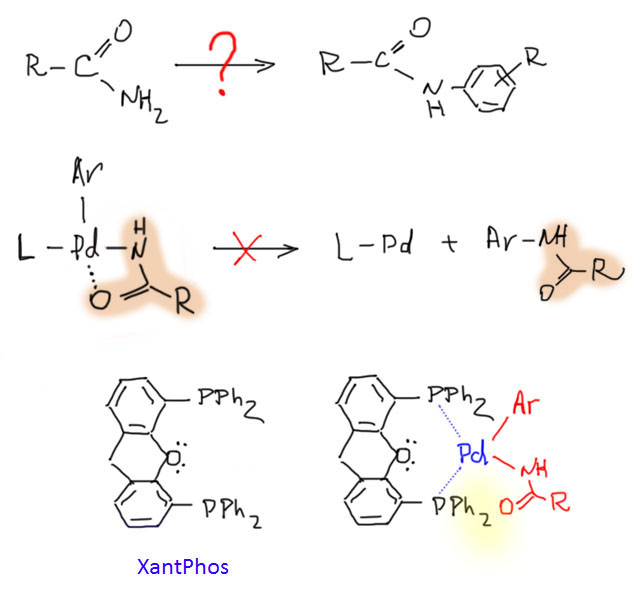
Еще одна проблема, которую долго не удавалось решить - непотребное поведение некоторых важных субстратов - и электрофильных и нуклеофильных реагентов кросс-сочетания - которые умеют цепляться за палладий не только тем атомом, на котором происходит кросс-сочетание и образуется новая связь, но и каким-то еще атомом, находящимся в месте, подходящем для хелатного связывания. Типичный пример такого субстрата - амид. Потребность в кросс-сочетании с амидами очень велика - среди амидов множество полезных молекул. Но с известными и уже отлично зарекомендовавшими себя лигандами в реакциях кросс-сочетания с аминами, амиды не реагировали. Исследования показывали, что они цеплялись за палладий еще и кислородом и активно сопротивлялись попыткам вышибить продукт реакции из координационной сферы. Реакция начиналась, но быстро глохла.
С проблемой удалось справиться подбором специальных лигандов, самым популярным из которых стал XantPhos, к услугам которого обычно и прибегают тогда, когда видят, что кросс-сочетание идет, но дает очень маленькие выходы, потому что продукт реакции плохо выходит из координационной сферы и блокирует обращение катализатора.
Занятно, что очень похожая проблема хорошо известна в ферментативном катализе - ингибирование фермента продуктом ферментативной реакции.
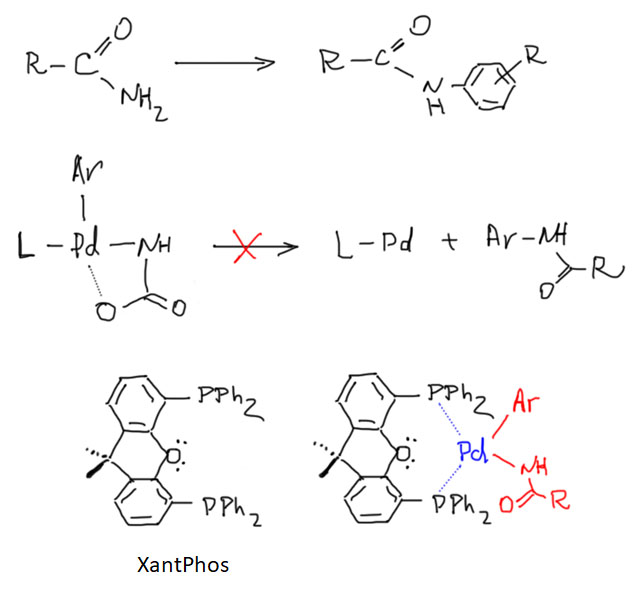
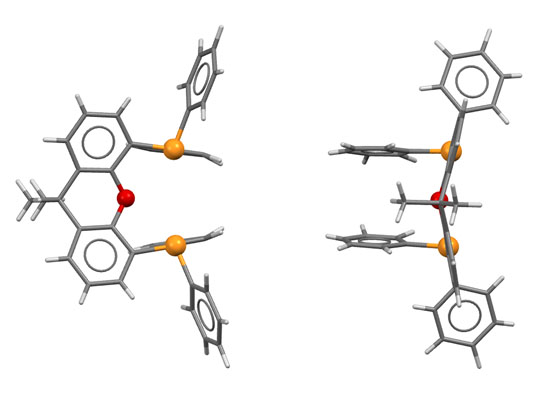
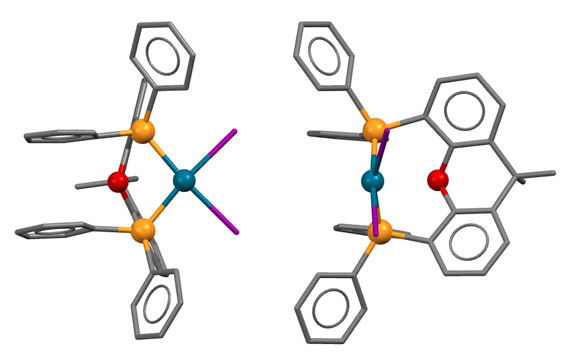
У КсантФоса, описанного довольно давно (Haenel et al, Tetrahedron Lett., 1995, 36, 75-78), но долго не находившего достойного применения, фосфиновые центры смонтированы на молекуле ксантена (так можно не совсем целиком передать популярное в англоязычной литературе mounted on xanthene scaffold) и очень сильно разнесены в пространстве (расстояние P-P более 4.1 ангстрем, а это как раз и есть приблизительно двойное расстояние металл-фосфор). Формальный угол укуса этого лиганда практически близок к 180 градусам. Атом палладия фактически целиком входит в такую широко раскрытую хелатную пасть, так что может образовывать хелат с транс-расположением фосфоров. Такая возможность выглядит парадоксальной, потому что раньше считалось, что хелатирование всегда использует два соседних места в координационной сфере и дает комплексы цис-конфигурации.
Так ли это, и хорошо это или плохо для целей каталитической реакции?
XantPhos - лиганд, который умеет приспосабливаться. Он может быть координационно стабильным хелатором, а может быть гемилабильным, и освобождать одно координационное место для лигандов, участвующих в реакциях.
Основа (scaffold) лиганда, молекула ксантена не совсем плоская, она чуть-чуть изогнута книжечкой, и поэтому атомы фосфора оказываются немного повернуты в пространстве в одну сторону. Это позволяет им быть в некоторых случаях цис-хелаторами, а в других - транс-хелаторами, переходя из одного типа связывания в другое. Примечание: желтые шарики - атомы фосфора, красный - кислород.
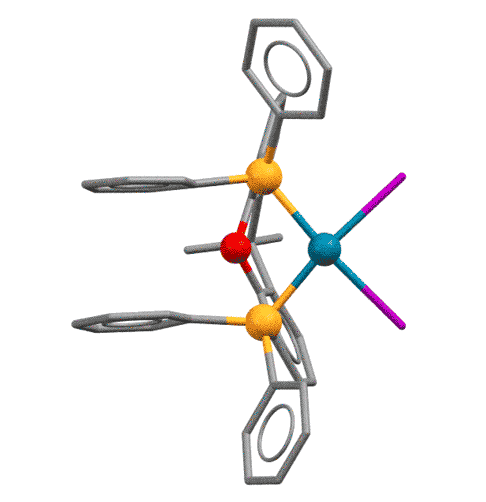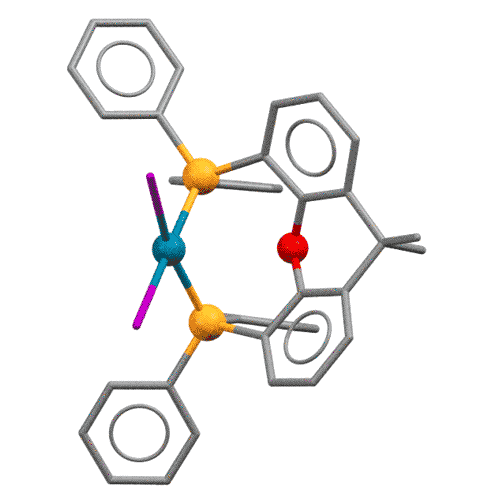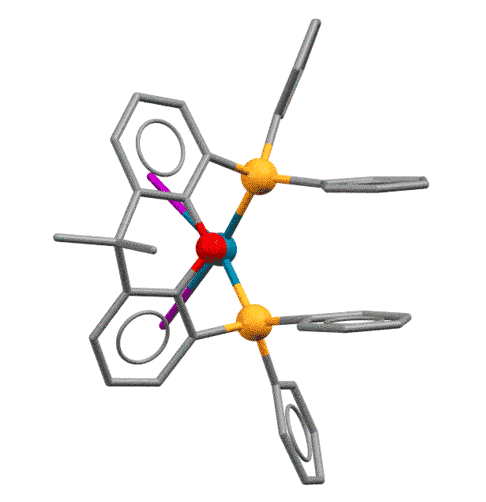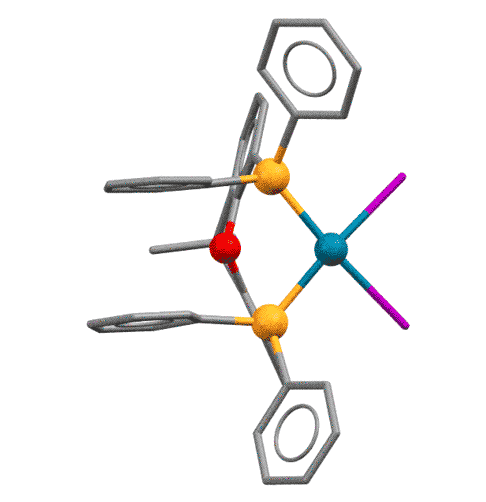
Grushin, Angew. Chem. Int. Ed. 2012, 51, 3668–3672
Grushin, J. Am. Chem. Soc., 2006, 128, 12644-12645
Кроме реакции C-N кросс-сочетания, которая произвела настоящую революцию в синтезе соединений азота, чрезвычайный интерес представляет открытая практически одновременно реакция образования связей углерод-бор, которую принято называть борилированием. Сочетание этой реакции с кросс-сочетанием Судзуки-Мияуры исключительно удобно. и очень сильно расширяет возможности этого метода, фактически открывая дорогу для кросс-сочетания двух разных галогенпроизводных или трифлатов без необходимости предварительного получения борных кислот.
Практически одновременно с C-N кросс-сочетанием Бачуолда-Хартвига была описана другая важнейшая реакция кросс-сочетания. Норио Мияура открыл реакцию, позволяющую из ароматических бромпроизводных или трифлатов фенолов получать борные кислоты напрямую.
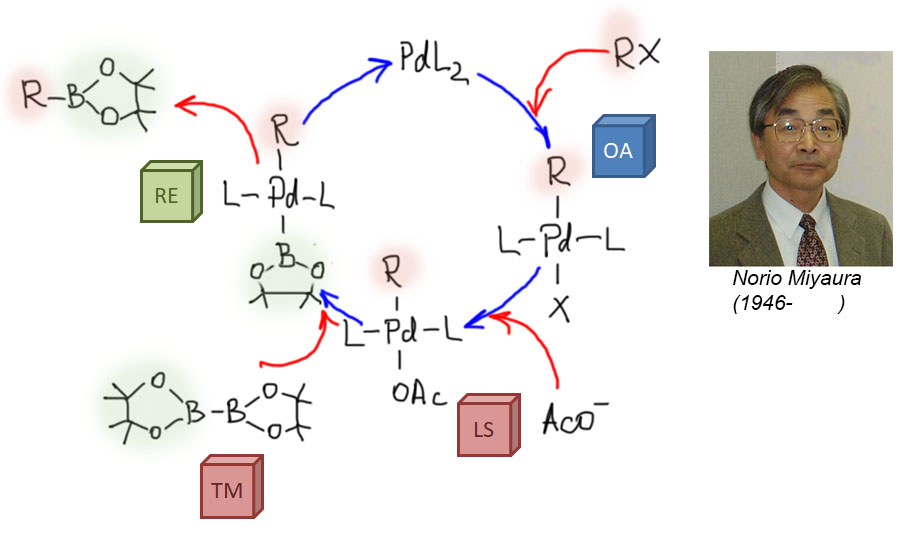
В этой реакции используется необычный нуклеофил - симметричное соединение со связью бор-бор, бис(пинаколил)диборан обеспечивающее загрузку борного остатка в результате переметаллирования. Образуются при этом не сами борные кислоты, а их пинаконовые эфиры, которые можно, но не так просто превратить в борные кислоты. Впрочем, сами эти эфиры вполне можно применять в реакции Судзуки-Мияуры, хотя иногда требуются изменения в методике - их реакционная способность немного ниже.

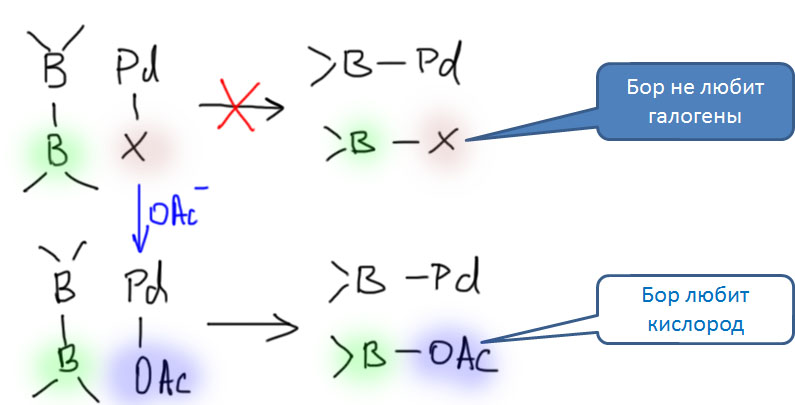
Реакция борилирования не потребовала такой кропотливой работы с подбором лигандов, как реакция C-N кросс-сочетания, но также потребовала точного знания механизма. В этом случае главным препятствием было то, что переметаллирование и вход борной части кросс-сочетания в координационную сферу никак не хотел происходить. Решение проблемы оказалось довольно простым и изящным - нужно просто заместить галогенид в координационной сфере палладия на кислородный лиганд типа ацетата или фенолята, и реакция сразу пошла.
Реакция идёт в присутствии специфических и довольно слабых оснований, ацетатов или фенолятов. Если взять основание посильнее, мы нарвёмся на конкурентную реакцию Судзуки-Мияуры - образующийся борный эфир будет вступать в реакцию с ещё непрореагировавшим субстратом-электрофилом и давать продукт кросс-сочетания, который в таком случае фактически является продуктом гомо-сочетания и никому, естественно, даром не нужен.
Реакция борилирования открывает очень важную возможность фактически осуществить кросс-сочетание электрофила с электрофилом (галогенпроизводного с другим галогенпроизводным, галогенпроизводного с трифлатом, трифлата с другим трифлатом). Один из субстратов превращаем в пинаколилборный эфир и вводим в реакцию кросс-сочетания.
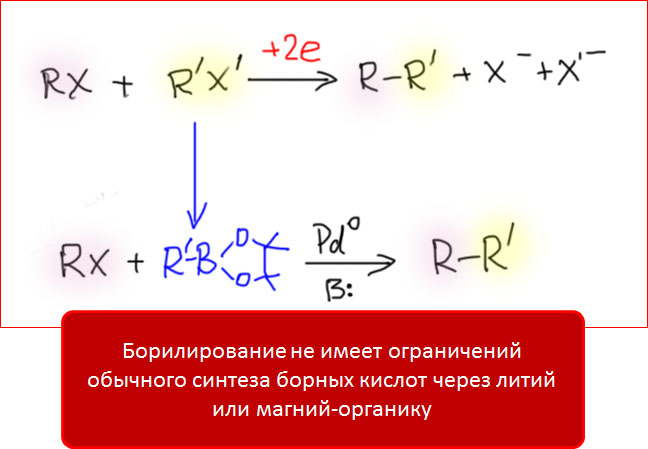
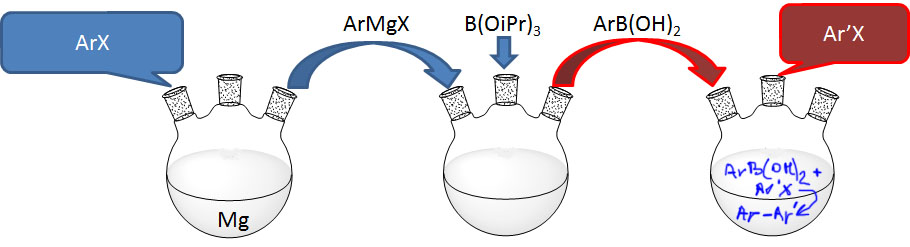
До открытия борилирования, та же задача решалась последовательностью трех реакций, каждая из которых делается отдельно в отдельном реакторе. Необходимость получения магний или литийорганического соединения еще и сильно ограничивает выбор одного из электрофилов.
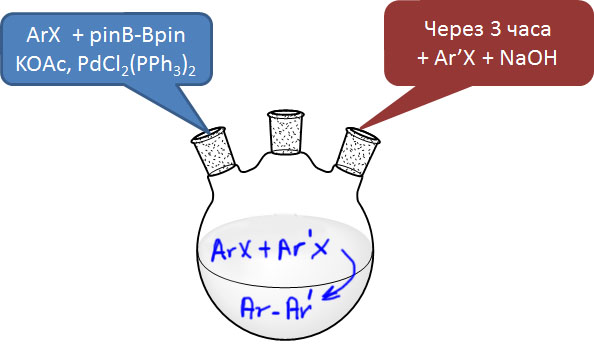
Реакция борилирования не только лишена этих ограничений, но и может быть сделана в том же реакторе, в том же растворителе, и даже с тем же комплексом палладия.
С практической точки зрения это выглядит как один синтез с последовательной загрузкой реагентов. Важно только не перепутать. Хотя - какая разница.
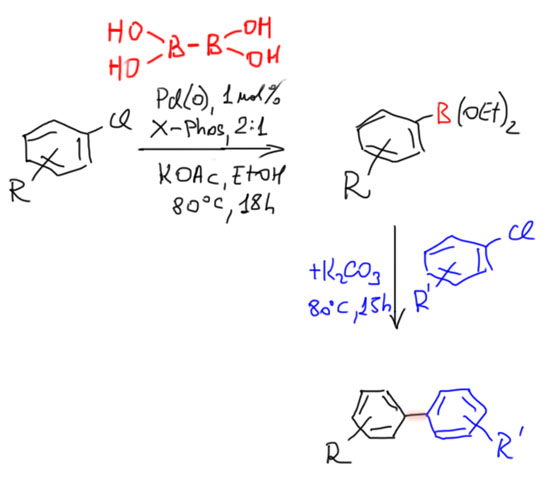
Главным недостатком борилирования по Мияуре является необходимость использования бис-пинаколилдиборана, довольно дорогого вещества, все части которого в результате идут в отходы реакций борилирования и Судзуки. Более того, пинаколиновые эфиры борных кислот, получаемые в результате реакции борилирования не всегда реагируют в реакции Судзуки так же хорошо, как сами борные кислоты.
Остроумное решение этой проблемы нашел Гэри Моландер с сотрудниками (G.Molander et al, J.Am.Chem.Soc., 2010, 132, 17701), использовав гораздо более дешевую гипоборную кислоту. Но, как мы уже неоднократно видели, упрощение и удешевление одних компонентов обычно достигается за счет усложнения других: система Моландера требует лиганды 3 поколения.
Мы говорим “кросс-сочетание”, подразумеваем “палладий”. Ну, иногда, один раз из ста, ещё никель. Очень долго это было непреложной истиной. Робкие попытки пристроить к делу другие металлы производили комическое впечатление ничтожностью достигаемых результатов. Палладий-катализируемое кросс-сочетание фактически стало устойчивым словосочетанием, в котором невозможно было изменить ни одного слова.
А ведь кросс-сочетание, как мы успели заметить, исторически было связано не с палладием, а с медью. Но в конце 20 века палладий настолько прочно оккупировал эту область, что про медь забыли все. Оказывается, не все. И с середины 1990-х совершенно внезапно начинается возрождение меди в кросс-сочетании. И сейчас, спустя более двух десятилетий, видно, что это было не недоразумением, а устойчивой тенденцией. За это время медный катализ в кросс-сочетании отвоевал себе настолько впечатляющий кусок этой химии, что больше говорить о монополии палладия решится только тот, кто совершенно утратил связь с современными достижениями в химии. Более того, как любят говорить вояки, “на плечах” меди в область вторглись и другие металлы, и сейчас в кросс-сочетании шумит совершенно удивительный и красочный карнавал множества металлов и их не менее живописных комплексов. Где ты, прекрасное далёко, когда всё было так ясно и понятно, что можно и что нельзя, и какой метод применить, а куда вообще не соваться? Сейчас можно всё, но вопрос как упирается в то, насколько мы верим в сотни новых протоколов катализа, практически не проверенных реальным синтезом. Поживём, попробуем, увидим.
Поэтому, так далеко мы пока в это углубляться не будем, но про медь вспомним…
Удивительно, но одна из самых первых (а возможно и самая первая) органическая реакция, катализируемая производными переходных металлов, была открыта в самом начале XX века (1901-1906) швейцарскими химиками Фрицем Ульманном и Ирмой Гольдберг (поэтому не называйте эту реакцию реакцией Ульманна-Гольдберга, как делают в нашей литературе почти всегда). Ирма Гольдберг - одна из первых женщин в химии, к тому же наша соотечественница, хотя ей и пришлось, как и многим, чтобы реализовать свои таланты, перебраться в другую страну. Эта именная реакция забавна ещё и тем, что это единственная семейная реакция, хотя Фриц и Ирма официально вступили в брак после публикации этих, без преувеличения, исторических работ.
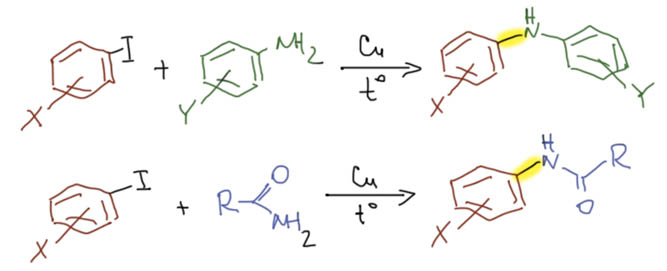
Ульманн описал арилирование ароматических аминов и фенолов ароматическими иодпроизводными. Реакция идёт в присутствии мелкораздробленного порошка меди (в старой литературе его обожали называть ульмановой бронзой) и требует весьма жёстких условий - длительного нагревания пр температуре за 200 градусов. Гольдберг одновременно описала арилирование амидов в очень похожих условиях.
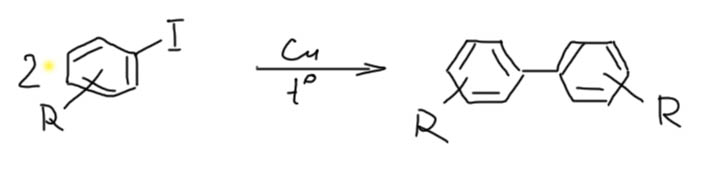
Ульманну также принадлежит метод получения дифенилов реакцией иод- или бромпроизводных с той же медью в столь же жёстких условиях. При внешнем сходстве с реакцией Ульманна-Гольдберг это не кросс-сочетание и не каталитическая реакция, а востановительное гомо-сочетание - в реакции участвуют два электрофила.
Реакция Ульманна-Гольдберг так и оставалась почти без изменений без десяти лет целый век. Время от времени её немного улучшали, пробовали разные формы меди и её простых производных (галогенидов, оксидов и т.п.), полярные и координирующие растворители типа хинолина и ДМФ, но ничего принципиального не происходило - реакция шла только при высоких температурах, была сильно ограничена в выборе реагентов, а её реальные применения в синтезе были эпизодическими и довольно простыми.
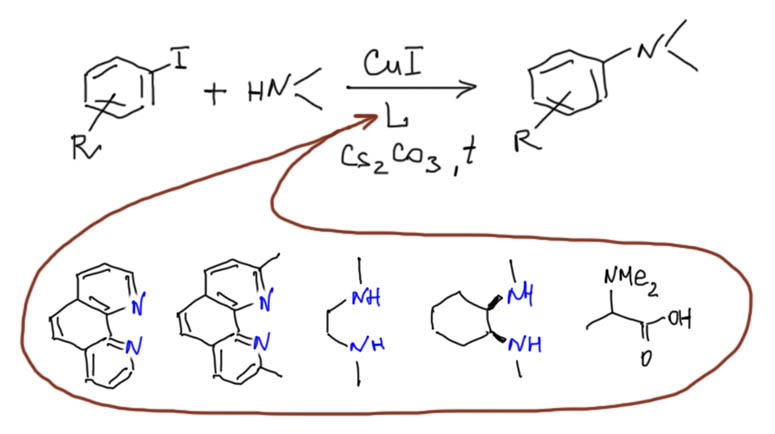
И вдруг, совершенно неожиданно, но по стечению обстоятельств почти одновременно с первыми работами по синтезу аминов Бачуолда и Хартвига появляются несколько работ, в которых показывают, что проблема решается издевательски просто добавлением к простой соли меди(I) очень несложных хелатных лигандов.
После этого реакция становится значительно более универсальной по отношению к природе азотного нуклеофила - классическая реакция ограничивалась ароматическими аминами, новая - вполне позволяла использовать и алифатические, и циклические, и ароматические. Реакция идёт в присутствии несложных оснований (карбонат цезия предпочитают по причине более высокой растворимости в органических растворителях). Условия реакции стали намного мягче, что сразу показало ее потенциал в сложном органическом синтезе.
С этого момента началась довольно жестокая гонка, в которой палладий - и медь-катализируемые реакции C-N кросс-сочетания стали быстро развиваться, оспаривая друг у друга первенство в синтезе азотсодержащих органических соединений.
Предполагаемый каталитический цикл медь-катализируемого кросс-сочетания принадлежит к другому типу циклов кросс-сочетания. Стадии окислительного присоединения и лигандного обмена поменялись местами - сначала в координационную сферу входит нуклеофил, и только потом электрофил. В отличие от нульвалентных металлов 10 группы, металл 11 группы в состоянии +1, формально имеющий ту же конфигурацию, обладает значительной льюисовой кислотностью и способен связывать нуклеофил.
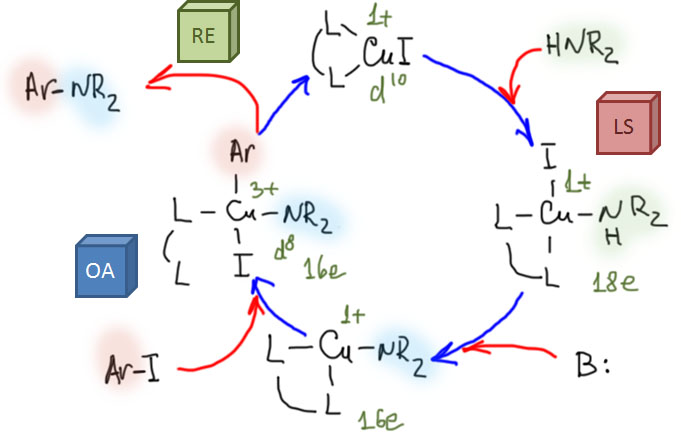
После депротонирования азотного нуклеофила в координационной сфере образуется донорный амидный лиганд, облегчающий окислительное присоединение к арилиодиду. Образующийся комплекс Cu(3+) быстро востановительно элиминирует продукт. Состояние Cu(3+) - сильный окислитель, поэтому здесь нет тех проблем, которые есть в палладий-катализируемой реакции, требующей специальных лигандов для "выпихивания" продукта из координационной сферы.
В отличие от механизма (каталитического цикла) палладий-катализируемого кросс-сочетания, очень тщательно исследованного и практически доказанного экспериментально, каталитический цикл медь-катализируемой реакции в существенной мере гипотетический. Наиболее спорным в этом механизме является участие Cu(3+) - такое состояние меди известно и присутствует в нескольких комплексах, но очень специального вида. Поэтому для реакции предлагались и альтернативные механизмы, не требующие образования Cu(3+).
Довольно долго было непонятно, что же делают лиганды в этой реакции. Спустя несколько лет после ее открытия были найдены многие десятки пригодных лигандов, от простых до очень простых. Среди них в основном хелаторы, но есть и совершенно примитивные монодентатные лиганды. Первоначальная гипотеза о необходимости образования специальных комплексов с фиксированной структурой не оправдалась.
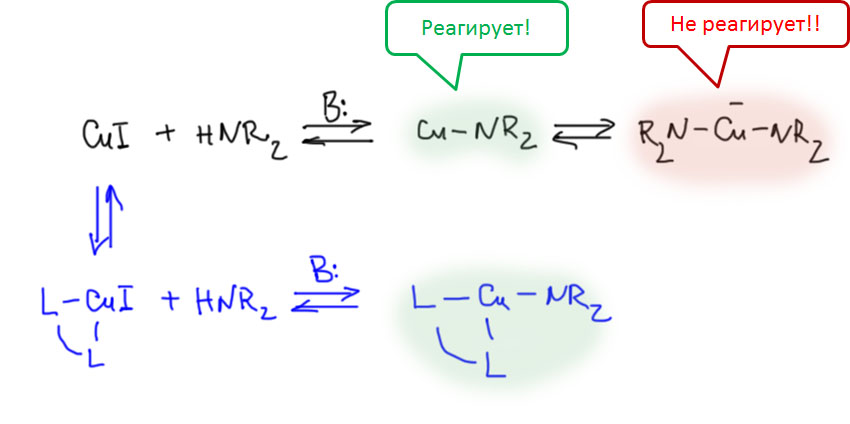
Наиболее вероятный механизм действия лиганда очень прост - он не даёт азотному нуклеофилу входить в координационную сферу в избыточных количествах, образуя неактивные в окислительном присоединении комплексы. Хороший хелатный лиганд сразу занимает два места у атома меди, и позволяет связать ровно одну молекулу азотного нуклеофила.
Получается, что результат реакции зависит от соотношения констант связывания анциллярного лиганда и нуклеофила. Поскольку это изменяется в очень широких пределах, это объясняет, почему подбор лиганда требуется почти под каждый новый нуклеофил, и никаких общих правил нет. Это довольно неудобно, и представляет собой серьёзный недостаток метода. Но поскольку все лиганды для этой реакции очень дешевы и доступны, а в литературе уже накопилось множество работающих протоколов, метод всё равно выгодно отличается от реакции Бачуолда-Хартвига дешевизной и простотой. Но - только там, где работает. Палладий-катализируемая реакция была и остаётся основным методом из-за гораздо большего диапазона.
Простота и дешевизна медь-катализируемых протоколов образования связей C-N (а также C-O, C-S и других) привлекает к этой химии огромное внимание. Но методы не лишены весьма серьёзных недостатков.
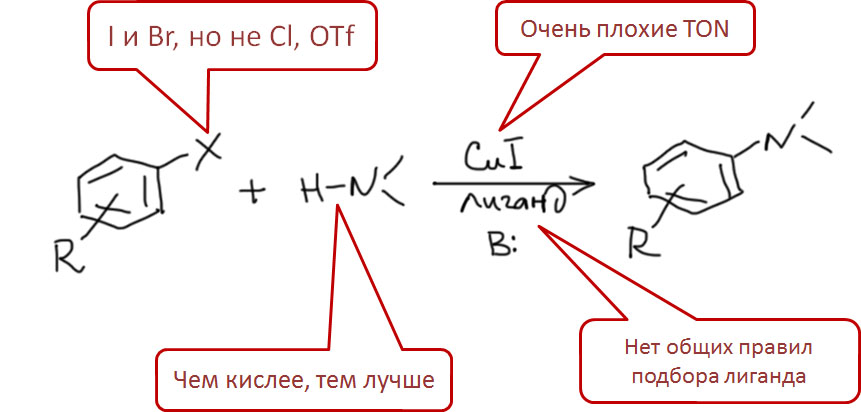
Очень ограничен выбор уходящих групп практически только тяжёлыми и дорогими галогенами.
В реакциях обычно требуются очень большие количества солей меди - мало каталитических циклов (часто не более 10)
Довольно ограничен выбор NH-нуклеофилов: эти реакции гораздо чувствительнее к стерике, чем палладиевые, и лучше работают с более кислыми NH-кислотами.
И возможно самое главное - слишком много лигандов и нет правил выбора хотя бы типа лигандов, и это требует перед применением реакции в новом случае проводить оптимизацию, то есть целую научную работу.
Но от меди не только не отказываются, но и продолжают работать над совершенствованием. В последние 10 лет в этой химии очень много новых работ. Со временем мы обратим на них внимание.