
Комплексы малых аннуленов
Циклопентадиенильный анион в качетве лиганда известен всем. А как поживают другие ароматические ионы, которых не так мало. Среди них есть и катионы (циклопропенилий и тропилий) и анионы, точнее, дианионы, производные циклобутадиена и циклооктатетраена. Есть ли комплексы таких ароматических соединений, и если есть, то что делать с теми, которые в свободном виде положительно заряжены, ведь всё-таки мы мыслим лиганд, связанный через атомы углерода, скорее как анион, чем как катион.
Да, такие комплексы есть и их много, они довольно популярны в координационной химии переходных металлов, и понемногу находят своё место и в катализе. Но для нас сейчас даже более интересно, что рассматривая такие комплексы, мы сможем вдоволь позабавиться с изученным материалом – с гаптностью, back-donation, счётом электронов, степеней окисления и т.п.
Напомню, что ароматических ионов не так уж и много. Вот почти полный список известных моноциклических ароматических ионов, не имеющих гетероатомов в цикле, а к комментариям по этому ряду отсылаю вас к моему органическому сайту. Все это симметричные правильные плоские многоугольники, вполне годные для того, чтобы стать лигандами в сэндвичевых (типа ферроцена) или полусэндвичевых (типа цимантрена) комплексов переходных металлов. Важно еще и то, что число электронов в π-системах различно – от двух до 10. Десятиэлектронные лиганды представляют проблему для комплексов – слишком много электронов сразу, и если у нас металл имеет конфигурацию d8, уже одного металла и одного лиганда достаточно для 18 электронов. Комплекс, аналогичный ферроцену имел бы 26 электронов – беспрецедентно много, столько не бывает никогда. И даже металл с конфигурацией d0 имел бы в таком сэндвиче 20 электронов, тоже многовато, хотя такие комплексы иногда встречаются, но обычно бывают малоустойчивы. Сразу понятно, что в комплексах металлов есть проблема, и фантазии разгуляться не получится. С 14-, 18– и более электронными ароматическими аннуленами можно распрощаться сразу – столько электронов металлу не переварить, даже если абстрагироваться от другой важной вещи – геометрических размеров таких систем. Это ферроцен выглядит со стороны как гамбургер между двумя булочками, а если бы мы представили комплекс металла с большой ароматической системой, металл выглядел бы как вишенка на торте или прыщ на широком лбу мыслителя, и нормального взаимодействия орбиталей металла и лиганда просто не получилось бы. 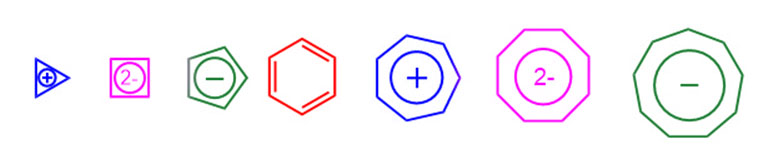
Циклопропенилий
Самый простой, а точнее, самый маленький ароматический циклический ион – циклопропенилий. С этого иона в послевоенные годы началась ароматическая лихорадка – гонка между несколькими амбициозными исследователями за приоритет в получении новых ароматических соединений, предсказанных теорией Хюккеля еще в 1930-х, которая в то время осталась практически незамеченной, потому что никто сразу не осознал, что какая-то математическая формула, выведенная молодым физиком, никогда до этого не имевшего никакого отношения к настоящей химии, химии соединений, а не математических формул (первой работой молодого Эриха Хюккеля была разработака теории электролитов вместе с голландцем Петером Дебаем – Дебай за это получил нобелевку, признался в большой любви и уважении к фюреру немецкого народа, получил место в престижном институте кайзера Вильгельма в Берлине, немного поработал в нацистской столице и успел хорошенько замараться, но смекнул прямо под ленточкой Второй Мировой сделать благодетелям ручкой и свалить от греха подальше в 1939 в США, где и осел. А его молодой ассистент Эрих Хюккель, судя по всему, не менее, а намного более талантливый, чем его прыткий голландско-нацистский начальник, остался в Германии, чтобы разделить плачевную судьбу несостоявшегося тысячелетнего райха. Здесь обойдёмся без подробностей. но когда-нибудь я подробнее расскажу эту поучительную историю про то, как добросовестный и совестливый человек невольно связывает свою судьбу с чудовищным режимом, рассказывая самому себе, что он работает на родину, а не режим. Но перед самым началом нацизма успел сделать несколько теоретических работ, которые навсегда обессмертили его имя. В одной из них он и вывел математическую формулу, описывающую положение и относительные энергии молекулярных орбиталей в π-системах циклических сопряженных симметричных системах, которые впоследствии получили название аннуленов. Его целью было показать причину ароматичности бензола, а заодно и поразительного поведения другой известной в это время частицы – аниона циклопентадиена, про которые Эриху рассказал его брат Вальтер, настоящий органический химик (и, кстати, очень недурной, – я много пользовался в своей собственной работе одной его методикой и признаю, что она превосходна, и сэкономила мне много времени и сил). Формула тогда мало кому показалась великим открытием, и так и пролежала под спудом, пока после войны на неё не наткнулось сразу несколько исследователей, понявших, что в ней, возможно, заложена просто невероятная предсказательная сила, стоит только поискать другие соединения, которые могут удовлетворять условиям Хюккеля, которые мы нынче знаем, как критерии ароматичности. Забавно, что сам Хюккель тогда был жив и здоров, но не только ничего уже не мог добавить к своему открытию, но и попробовав сам как-то продолжить исследования, быстро сдался, поняв, что за годы нацизма полностью потерял и квалификацию, и интерес к иследованиям, да и просто безнадёжно отстал от мировой науки – Германия была практически отрезана от научного обмена и нормальной научной конкуренции практически с самого 1933-го года, так как сначала фюрер указал, что немецкая наука – самая научная наука в мире, и никто ей не нужен, а всех несогласных и недостаточно патриотичных а заодно и всех, у кого была хоть капля еврейской крови – быстро из науки выперли, ну а потом и вовсе стало как-то не до фундаментальной науки, и наукой стало считаться только то, что способствовало бы победам немецкого оружия. Как обычно в таких случаях бывает, дорожка от побед к поражениям оказалась не очень долгой.
Первым как раз и был циклопропенилий, ароматический ион, впервые полученный Роном Бреслоу в конце 1950-х (Breslow, R. J. Am. Chem. Soc. 1957, 79, 5318). Очень полезно это осознать – от теории Хюккеля до этого первого объекта, предсказанного этой теорией, прошло почти 30 лет. После этого все буквально сошли с ума, бросившись искать других представителей, как тогда любили говорить, небензоидной ароматичности. И тогда же был получен ферроцен, и установлена его структура. И доказана структура дибензолхрома. Немедленно возник вопрос – а есть ли на свете похожие на ферроцен и дибензолхром комплексы с другими ароматическими системами.
Первый комплекс циклопропенилия был получен немного случайно в работе, посвященной аллильным комплесам. Был получен аллильный комплекс молибдена и, как аналог, комплекс трифенилциклопропенилия с использованием точно такого же протокола (Hayter, R. G. J. Organomet. Chem., 1968, 13, P1 ) Здесь хорошо видно, что аналогия действительно полная – хлорпроизводное циклопропена в ковалентной форме это типичное аллильное галогенпроизводное. Хлорид – анион с немаленькой нуклеофильностью, поэтому это ковалентное соединение, а не соль циклопропенилия, как было бы, если бы анион был менее нуклеофилен. Но в комплексе циклопропенилий – плоский симметричный η3-лиганд, что определилось по симметричности резонансных линий в ЯМР-спектре. И лиганд сидит прочно, так что удалось туда же затащить еще и обычный Cp.
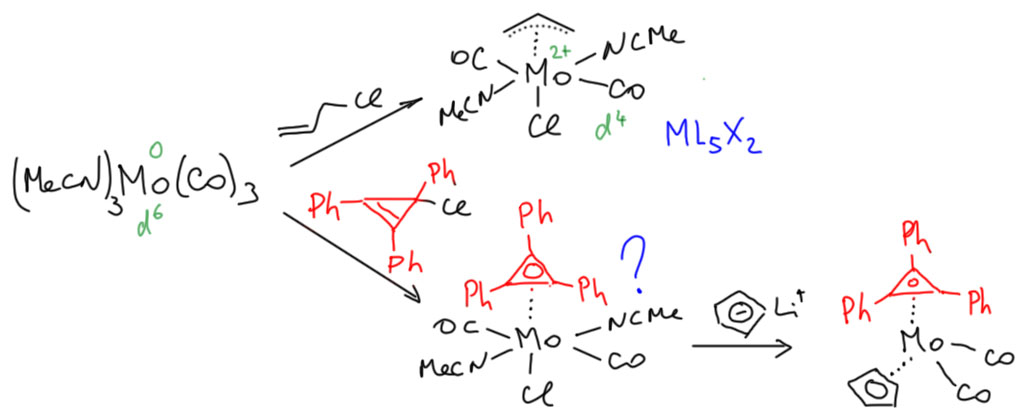
Только напрашивается вопрос – а как представлять такой лиганд? Как XL – по аналогии с аллилом – ведь комплексы изоструктурны. Тогда и во втором комплексе молибден имеет степень окисления 2+? А как это совместимо с тем, что мы хотели бы, чтобы этот лиганд был ароматическим циклопропенилиевым катионом? И если это ароматический двухэлектронный лиганд, то сколько электронов он дает металлу – два или четыре? Если четыре, как аллил, то где он их берёт? А если не четыре, то почему структурные типы одинаковы? Или это иллюзия? Придётся попробовать разобраться, и заодно потренироваться.
Так что за лиганд этот циклопропенилий?
Вместо ковалентного производного циклопропена стали брать готовые соли ароматического катиона циклопропенилия, обычно с тремя крупными заместителями, фенилами или трет-бутилами. И такие соли реагируют с нуклеофилами вполне в рамках обычного механизма SN1 c образованием замещённых циклопропенов. Хотя мы еще не проходили нуклеофилы на основе переходных металлов, поверим на слово, что такие есть, и очень хорошие. Вот к чему приводит реакция одного такого знаменитого нуклеофила, анионного комплекса железа с циклопропенилием. 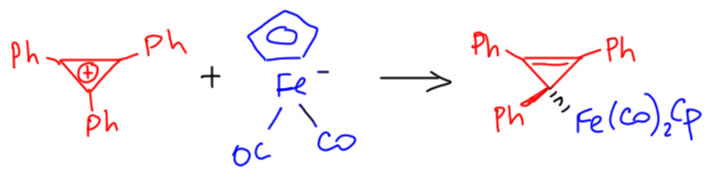
И вот структура комплекса – это обычный σ-комплекс, в котором циклопропенил сидит как обычный X-лиганд, железо имеет степень окисления 2+, и вообще ничего необычного не случилось. Вот структура комплекса (Gompper, R. et al. Chem. Ber. 1979, 222, 218). Железо в нём координационно насыщено, имеет структурный тип FeX2L4 и 18 электронов. Очевидно, что зацепиться за циклопропенил ему больше нечем. Так и болтается. Вполне соответствует обычным моногапто-аллильным комплексам.
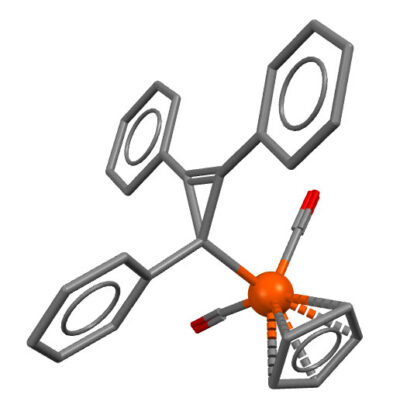
А вот очень похожая реакция, циклопропенилий реагирует с нуклеофильными анионами из 9 группы (R. P. Hughes, D. S. Tucker and A. L. Rheingold, Organometallics, 1993, 12, 3069). И в этой реакции, точнее, реакциях, получаются η3-комплексы, в которых трехчленный цикл сидит симметрично, как мы и хотели бы ожидать от ароматического иона. И если судить по стехиометрии – катион реагирует с анионом с образованием нейтрального комплекса – то получается, что в комплексе металл не изменил степень окисления, она осталась -1, а лиганд ведёт себя, как 2-электронный тригаптный донор – то есть структурный тип здесь ML4–, такой же как в исходном анионном карбонилатном комплексе (минус нам нужен, потому что при таком подходе лиганд положительно заряжен, а нам нужно получить общий заряд 0). При таком подходе циклопропенилий просто заместил карбонил в комплексе. Но посмотрим на процесс с другой точки зрения, разложив его по стадиям, как показано внизу на желтоватом фоне. Тогда сначала происходит точно такая же реакция, как мы уже видели выше, при этом металл меняет степень окисления на 2 единицы. А после он теряет CO и связывается с двойной связью обычной донорно-акцепторной связью. Результат – тот же комплекс, мы попали туда же, но по дороге изменили степень окисления металла. И структурный тип – теперь он MXL4. И теперь мы видим тот же комплекс, но трехчленный цикл уже XL лиганд, причём четырёхэлектронный. О, боги, откуда же он взял вторую пару электронов, ведь это же была 2-электронная ароматическая система!! Эх, вы плохо следили за руками – эта пара незаметно была подсунута металлом, как заправским напёрсточником – ведь при образовании моногапто-комплекса вон там внизу слева на желтоватом фоне именно металл даёт пару на образование связи – это типичный back-donation, как иногда говорят, дативная связь (давайте не привыкать к этому странному термину, но приходится признать, что в литературе он встречается).
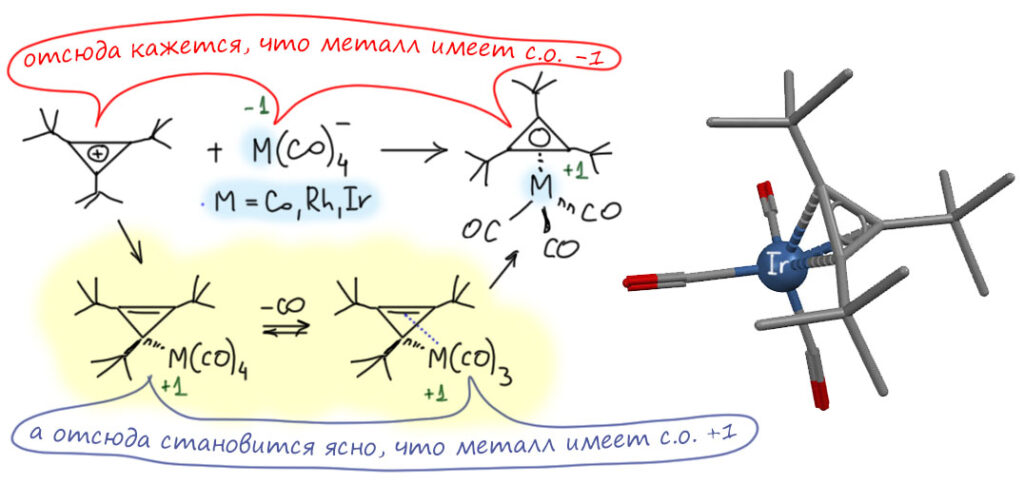
На вашем месте я бы устроил скандал – так как же правильно? Что за бардак! Так циклопропенилий это 2-электронный или 4-электронный лиганд? А у металла степень окисления какая?
Спокойно, пора уже привыкнуть, что в тех случаях, когда в образовании комплекса очень большую роль играет back-donation, а это явно очередной такой случай, мы действительно имеем возможность смотреть на комплекс двумя способами, и и металл имеет неопределенную степень окисления – можно так, а можно и по другому, важно только определиться, какой способ мы предпочитаем. Не забывайте, что степень окисления – это формальная величина, а не объективно измеряемое свойство. Степень окисления зависит от того, как мы договорились смотреть на лиганды и образование связей. И обратите внимание, что как бы мы ни посмотрели, получаем нечто разумное, а такая вещь, как счет электронов не зависит от способа разложения комплекса на формальные части. А счет электронов – штука довольно важная, потому что служит для оценки координационной насыщенности.
И ещё одно соображение. Когда мы решили считать циклопропенилий XL-лигандом, мы фактически мысленно преобразовали его из катиона в анион с помощью пары, впрыснутой металлом с помощью back-donation. И не надо возмущаться, что циклопропенильный анион антиароматичен. Мы скоро доберемся до циклобутадиена и поймём, что лиганд не подчиняется правилу Хюккеля в буквальном смысле этого правила. Но есть и ещё один способ посмотреть на этот лиганд в таких комплексах, и не устраивать эту малость мошенническую операцию – берем пару у металла и считаем ее парой лиганда. Можно и сотавить ее у металла, если мы будем считать циклопропенильный тригапто-лиганд лигандом не XL, а ZL-типа именно потому что вторая пара всё же идет от металла. В этом случае, металл опять становится M(-1), а лиганд двухэлектронным.
Вот какая запутанная ситуация. И все три подхода имеют право на существование и при аккуратном учете электроно дают одинаковые результаты.
Циклобутадиен
Хотя мы здесь решили заниматься ароматическими ионами, и нужно было бы обсуждать дианион циклобутадиена, но химия переходных металлов устроена так, что никогда заранее не знаешь, в каком состоянии находится лиганд. Поэтому начнём именно с циклобутадиена. Все знают, что это антиароматическое соединение, и что хотя сам циклобутадиен в обычных условиях неустойчив, его комплексы с переходными металлами известны. Но не так просто понять, что все это значит, и почему антиароматическое соединение вдруг начинает чувствовать себя отлично в составе каких-то комплексов. Тут есть даже некая несправедливость – ведь мы так радуемся ферроцену и дибензолхрому, а также другим комплексам этих систем; тому, насколько эти соединения стабильны и как хорошо они вписываются в общую теорию ароматичности. А у нас просто не возникает сомнения в том, что причиной такой поразительной стабильности и проч. является именно ароматичность.
Но что тогда с комплексами циклобутадиена? Эта молекула в виде лиганда почти всегда представляет собой плоский квадрат – тот самый квадрат, от которого убегает, как чёрт от ладана, свободный циклобутадиен. И комплексы эти вполне стабильны, может не совсем настолько, насколько стабилен ферроцен, который, кажется, способен выдержать прямое попадание ядерной бомбы (но только прямое, ведь если ядерна бомба взорвётся рядом, то ферроцен сдует ударной волной, мы его не найдём и не сможем узнать, выжил он или нет). Но вот с дибензолхромом многие комплексы циклобутадиена по стабильности сравнятся, да и даже превзойдут. Более того, многие комплексы циклобутадиена отлично реагируют в реакциях электрофильного ароматического замещения – дейтерирования, ацилирования, меркурирования, и т.п. Ну и что там с антиароматичностью? Может в комплексе он ароматическим становится? Или вообще лучше про ароматичность забыть, когда речь идёт о комплексах таких лигандов? Попробуем разобраться, это любопытная история, не вполне однозначная, но вполне содержательная.
Напомню, что такое циклобутадиен, и в чём его проблема. Это простейший антиароматический аннулен. Проблема всех антиароматических аннуленов, по теории Хюккеля, проста – граничными орбиталями у них является дважды вырожденная несвязывающая орбиталь. А поскольку на ней надо разместить пару электронов, то по всем правилам получается по электрону на каждую из вырожденных. Это очень нехорошая ситуация в структурной химии, и из неё следуют разные плохие вещи: а) что в совсем симметричном виде, например, как здесь, в виде правильного квадрата, эта молекула вообще неустойчива и не имеет шансов жить; б) что небольшой шанс жить у нее появляется, если из квадрата она исказится в прямоугольник так, что две связи станут короче и будут ближе к двойным а две длиннее – ближе к одинарным, тогда и вырожденная орбиталь разъедется на две с разной энергией, и два электрона спарятся на нижней. Но этот выход дает молекуле шанс жить только при низкой температуре, птому что такое искаженние не очень сильно разведет две орбитали, и между занятой и пустой будет очень небольшой зазор, а это еще одна скверная ситуация – в этом случае очень легко происходит возбуждение, переход одного электрона на орбиталь повыше, а это и есть то самое состояние, от которого мы ушли. В реальности это приводит к тому, что молекула не живет в человеческих условиях, и проявляет совершенно бешеную реакционную способность, например, вступает в реакции типа Дильса-Альдера со всем, до чего может дотянуться, а если ни до чего не может, то просто со второй такой же молекулой. Подробнее про то, что такое ароматичность, и что за проблема у того же циклобутадиена можно почитать на моем органическом сайте в разделе Ответы.
Вот что такое, вкратце, циклобутадиен. Не жилец. Это и есть, очень грубо говоря, зримое воплощение антиароматичности. Напомню также, что ароматическая система (аннулен) отличается от антароматической тем, что граничные орбитали у неё всегда связывающие и полностью заполненные, и лежащие очень далеко по энергии от пустых антисвязывающих.
Итак, вспомним как выглядят молекулярные орбитали бутадиена, π-орбитали естественно только, так как их даёт теория Хюккеля, удобно представляемая с помощью вписывания квадрата в окружность (мнемоника круга Фроста)
И вот антиароматический циклобутадиен попадает в состав комплекса с переходным металлом. Что при этом может произойти предсказал еще в 1956 году легендарный английский теоретик и основоположник метода МО Кристофер Лонге-Хиггинс. Это был совершенно гениальный ученый, один из первых, кто решил использовать квантовую науку для химии, и не так как Полинг – за счет потрясающего умения популяризации и профанирования – а по-возможности именно как теорию, при том как в то время никакие расчеты были недоступны. В этом и была его проблема – его работы были невероятно заумны, и химики их просто не понимали и не использовали.
Сильно упростив его выводы, можно сказать что
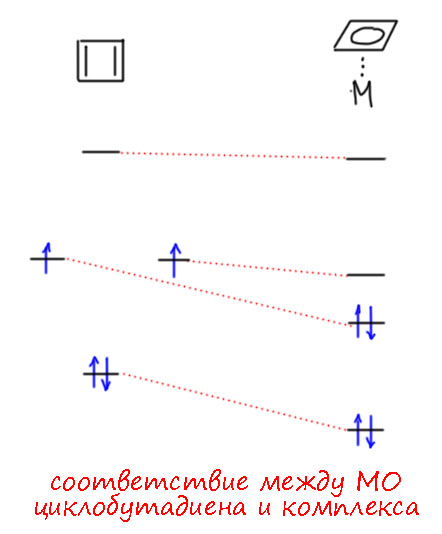
- в составе комплекса вырожденная граничная орбиталь квадратного циклобутадиена, скорее всего, перестанет быть вырожденной (не будем в этом подробно разбираться, но дело в том, что для вырождения нужна ось симметрии 4-го порядка, как в квадрате, а металл с другими лигандами, скорее всего, такую ось уничтожат своей более слабой симметрией)
- в результате взаимодействия с орбиталями металла, бывшая несвязывающая орбиталь, так сильно беспокоившая свободный циклобутадиен, станет связывающей – взаимодействие понижает энергию, стабилизирует.
- эта орбиталь станет источником пары электронов для обычной донорно-акцепторной связи с металлом. Другая пара возьмется с самой низкой занятой орбитали циклобутадиена, про которую мы как-то забыли, но она никуда не делась.
Смотрим на эти вырожденные орбитали. Сразу во избежание недоразумений поясню одно частое недоразумение. Вырожденными являются орбитали, имеющие одинаковую энергию. Если таких орбиталей две, их называют дважды вырожденными и очень часто допускают название дважды-вырожденная орбиталь, то есть как будто считают эту пару орбиталей одной двойной орбиталью. Это распространеннная терминологическая неряшливость – и не надо забывать, что орбиталей две, а не одна. При любом взаимодействии, которое снижает симметрию квадрата, например, при образовании комплекса с металлом, эти две орбитали разойдутся по энергии. А может быть так, что при образовании комплекса симметрия сохранится? Да, в принципе может, но такой комплекс представить себе довольно трудно. Я не видел ни одного. Было бы интересно посмотреть, что будет. Я думаю, если бы такой комплекс решил образоваться, он пожалел бы об этом, а в таких случаях обычно включается так называемый эффект Яна-Теллера, и высокосимметричный комплекс самопроизвольно исказится, и орбитали все равно разойдутся.
Какая из них опустится ниже? Любая из двух может. Это зависит от конкретного металла, его электронной конфигурации. По симметрии к этим двум орбиталям соответствуют две p-орбитали металла и две d-орбитали металла. Вполне достаточно. Не будем вдаваться в частности, просто посмотрим, что при взаимодействии с металлом бывшие дважды вырожденные орбитали снижаются по энергии, но на разную величину. Обе становятся связывающими. Пара электронов есть только на нижележащей. Очевидный вопрос – а почему мы рисуем орбитали циклобутадиена в комплексе – разве в новом соединении молекулярные орбитали не будут новыми, с участием и лиганда и металла? Конечно, именно так, именно такие новые орбитали мы и рисуем, но законы МО вполне однозначны – если мы делаем новое соединение из фрагментов (в данном случае из металла и лиганда), то имеем право в первом приближении комбинировать орбитали попарно, так чтобы новых орбиталей стало столько же, сколько было исходных. В этом случае, каждая орбиталь, например, лиганда переедет в орбиталь комплекса, и зваимодействие с подходящей орбиталью металла опустит ее по энергии, при этом образуется еще и новая антисвязывающая орбиталь. И не надо особенно комплексовать по тому поводу, что кто-то сделал расчет, и там все орбитали совсем другие. Это естественно, они просто по-другому скомбинированы, не попарно, а более сложной комбинацией всех подходящих по симметрии перекрывания. Но вот если мы просто посчитаем новые орбитали и заметим их тип и расположение по энергии, мы с удивлением увидим довольно хорошее соответствие прогнозам.
Итого – циклобутадиен сработал как обычный 4-электронный лиганд, в общем, как обычный диен, тетрагапто-лиганд типа L2. И где тут антиароматичность? Да нет её, говорить даже не о чем – стабилизация бывшей несвязывающей орбитали, наряду со снятием вырождения вообще убирает то, что и является в свободном лиганде причиной и признаком антиароматичности. Нет больше дестабилизации, ни по причине того, что электроны находятся на несвязывающей орбитали в состоянии вырождения, ни по причине того, что есть возможность легкого возбуждения с переходом одного электрона на следующую орбиталь. Про парамагнитность и другие признаки антиароматичности вообще говорить не будем, так как у нас теперь есть металл, и мы пока говорим все в общем виде и не знаем, какой точно металл, а металлы и сами бывает, что имеют неспаренные электроны и связанную с ними парамагнитность.
Но фокус в том, что это описание в общих чертах верно, но, возможно неполно. У нас есть одна проблема, о которой мы пока не говорили. Дело в том, что если в комплексе циклобутадиена все так отлично застабилизировалось, то циклобутадиеновый лиганд должен представлять собой плоский квадрат с делокализованной π-системой, похожей по структуре на бензол – в том смысле, что длины всех связей должны быть одинаковы и где-то в районе 1.40-1.42 Å. И всё вроде так – в большинстве известных комплексов это квадрат, да вот связи заметно длиннее – всё время что-то в районе 1.46 Å, но бывает и до 1.50 Å. О, боги, какая мелочь, стоит ли париться ради такой фитюльки?? Стоит. Длины связей – штука очень точная, и связи одного типа обычно воспроизводят длины с точностью до 0.02 Å. Так что это сильное отклонение, там уже и до совсем простой связи недалеко, собственно 1.50 это уже почти простая связь и есть, а чтобы из сопряженной пи-системы сделать простые связи, надо заселить все разрыхляющие орбитали циклобутадиеновой системы, на это нужно всего 8 электронов, но столько циклобутадиеновому лиганду никто не даст. А вот шесть дадут. Это разрыхляет связи в цикле, уменьшает порядок связей.
Собственно, совершенно очевидно, что происходит. Ведь когда в циклобутадиене лиганде мы сняли вырождение, отправили пару электронов на нижнюю и использовали ее для координационной связи с металлом, у нас осталась еще одна орбиталь, пустая. Причем эта орбиталь по определению лежит невысоко, ведь она осталась из той пары и никто ничего с ней пока не делал. Она просто просит электронов – дайте ей скорее – и мы знаем что иногда металлы умеют это делать ведь в этом и состоит эффект back donation. Мы можем сделать вывод, что циклобутадиеновый лиганд должен быть очень восприимчив к back-donation. Вот так дополним упрощенную диаграмму молекулярных орбиталей, нарисовав в явном виде атомные орбитали металла (схематически, не обозначая их тип, в некоторых случаях некоторые из них могут быть вырождены, но это зависит от симметрии комплекса, а у нас общий случай, симметрии нет, а значит нет и вырожденных орбиталей любого типа). Пустые орбитали металла пошли на координационные связи, а если найдется у металла хоть одна пара, ее можно пустить на back-donation, использовав вторую из бывших вырожденных орбиталей циклобутадиена Хюккеля.
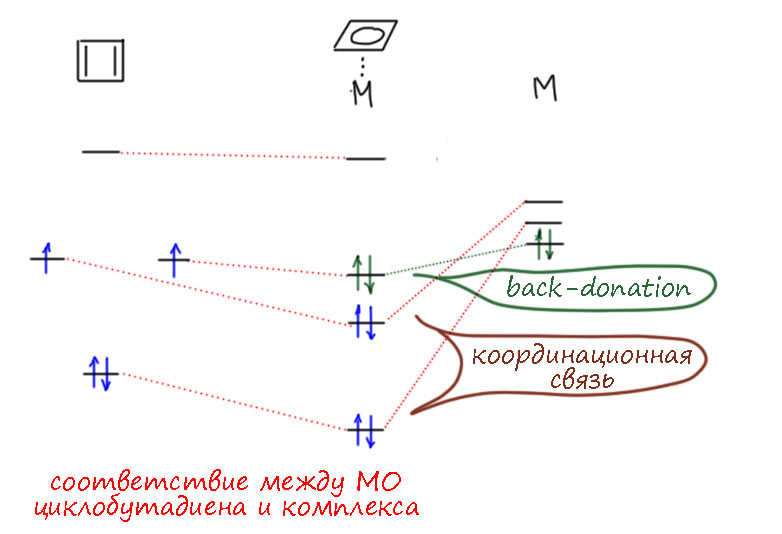
Что из этого может следовать:
- циклобутадиеновые комплексы скорее должны быть у низковалентных поздних переходных металлов, а если мы их найдём у ранних переходных металлов, то не в высшей степени окисления
- back-donation увеличивает электронную плотность на лиганде, а это закономерно увеличит длины связей, уменьшит порядки связей – но это мы уже видели
- циклобутадиеновые комплексы могут быть прочнее обычных диеновых того же лигандного состава именно потому что обычный диен не очнь нуждается в back donation, ведь та свободная орбиталь, которая может взять электроны у металла лежит намного выше – это настоящая разрыхляющая орбиталь. Back donation слегка разрыхляет сам лиганд, но упрочняет связь металла с лигандом.
Отсюда и вполне ясна склонность циклобутадиена в комплексах к электрофильному замещению.
Первый комплекс циклобутадиена не заставил себя долго ждать после работ Лонге-Хиггинса, но получен он был тоже немного случайно – немецкий химик, тоже из Мюнхена, Вальтер Хюбель с сотрудниками (Hübel W., Braye E. H. J. Inorg. Nucl. Chem. 1959, 10, 250) исследовали реакции дифенилацетилена с карбонилами железа и получили желтое кристаллическое и очень стабильное вещество с температурой плавления сильно выше 200 градусов. Структуру установили несколько позже другие исследователи (код структуры PCYFCO: R.P.Dodge, V.Schomaker, Acta Crystallogr. 1965, 18, 614), и это оказался комплекс тетрафенилциклобутадиена, лиганд оказался плоским квадратом: Это результат оказался весьма удивительным даже несмотря на предсказание Лонге-Хиггинса, мало ли что там заумные теоретики напридумывали, тем более в те годы химики еще не очень привыкли к тому, что в их священнодействие под тягам могут совать нос какие-то щелкопёры, в жизни своей даже хлорида натрия не получившие. Тем более, что сама реакция была необычна. Димеризация ацетиленов просто так не идет, и приблизительно в это время Вудвард и Хоффман показали почему. Даже наоборот, многие попытки до этого получить свободный циклобутадиен приводили к двум молекулам ацетилена в спонтанной циклореверсии. А тут такое!
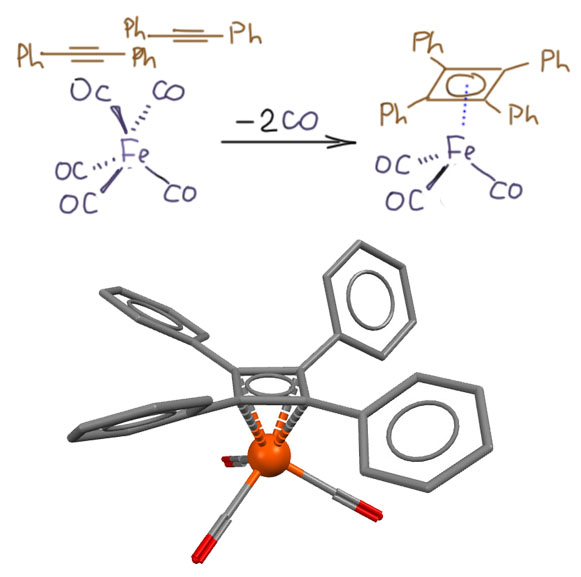
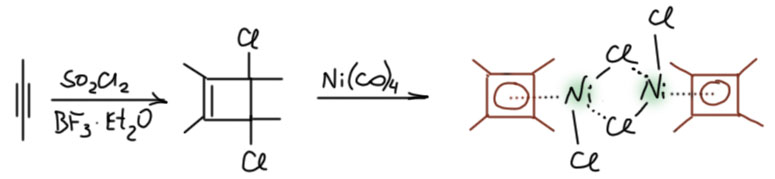
Четыре фенила могут как-то защищать циклобутадиеновое кольцо, поэтому сразу же захотелось получить циклобутадиен поменьше, настолько, что за тетраметилпроизводное взялся такой мощный химик как Рудольф Криге, основоположник озонолиза. Криге воспользовался очень интересным открытием украинского исследователя Ивана Смирнова-Замкова, который изучал хлорирование диметилацетилена, но получил совершенно необычный продукт, как будто аддукт хлора и тетраметилциклобутадиена. Криге немного усовершенствовал метод Смирнова-Замкова и попытался из этого соединения элиминированием получить сам тетрацетилциклобутадиен, но получил только димер. И тогда он сделал реакцию этого соединения с тетракарбонилом никеля, фактически осуществив окислительное присоединение (Criegee, R. Angew. Chem., Int. Ed. Engl. 1962, 1, 519). Надо отдать должное этому превосходному химику, хоть он однажды в 1943 и припёрся к нам с автоматом, но получил в физиономию хороший осколок и ретировался на родину лечиться и заниматься химией, что у него точно получалось намного лучше. Это ведь был химик еще старой немецкой школы, но он в отличие от большинства таких же классиков смело ринулся в новую химию, не побоялся новой металлоорганики, и в общем сделал одну из самых знаменитых своих работ. Получился комплекс никеля, как водится у элементом 10 группы, димерный, с мостиковыми хлоридами.
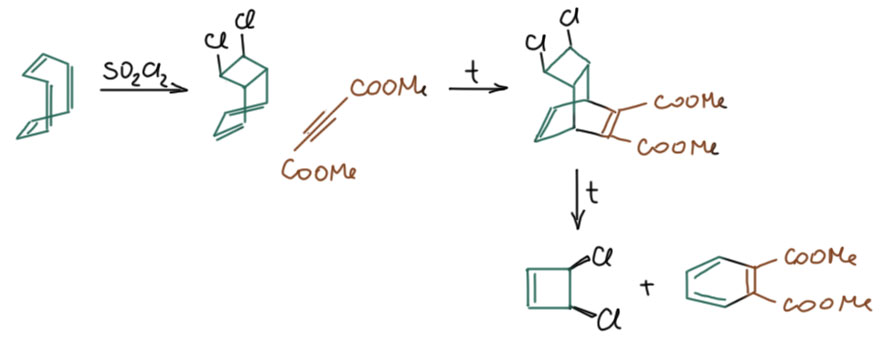
Но всё равно хотелось получить комплекс самого незамещенного циклобутадиена. За решение задачи взялся австралийский химик Роуленд Петтит (Emerson, G. F., Watts, L., Pettit, R. J. Am. Chem. Soc. 1965, 87, 131), который дальше вообще станет одним из главных по этому типу лигандов. Исходное заботливо подтащили румынские химики во главе с главным румынским химиком Костином Неницеску, обнаруживших весьма нетривиальный и быстрый способ наработки дихлорциклобутена в больших количествах из доступного промышленного циклоктатетраена. При частичном хлорировании циклооктатетрана замыкается циклобутановый цикл и образуется диен, который можно связать в реакции Дильса-Альдера с ацетилендикарбоновым эфиром, и дальше аддукт подвергнуть ретро-циклоприсоединению, причем образуется ароматический диметилфталат и дихлорциклобутен.
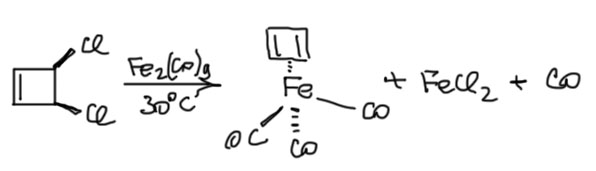
Этот дихлорциклобутен дальше использовали по методике Криге с разными низковалентными переходными металлами и получили много комплексов самого циклобутадиена. Самый первый, железотрикарбонильный, полученный Петтитом, оказался легкоплавкими кристаллами, вполне устойчивыми, что окончательно доказало, что заместители не нужны дляя стабилизации. После этого комплексы повалили десятками.
А вот альтернативный подход к циклобутадиеновым комплексам. Причем тем же самым. Рутений это аналог железа, комплексы имеют тот же структурный тип. Но получить их можно и тем же методом, каким получают комплексы с циклопентадиенилом – лигандным обменом с анионом. В данном случае использовали дианион циклобутадиена, это ароматическая 6-электронная система. Исходный комплекс для лигандного обмена – димер с мостиковыми хлоридами; учёт мостикового лиганда даёт нам структурный тип MX2L4 c Ru(2+) и это хороший координационно-насыщенный комплекс. Он реагирует – легко и чисто – с дианионом и образуется ровно то же самое, что мы только что видели у железа – комплекс с тремя карбонилами и циклобутадиеном. Там мы это интерпретировали как комплекс Fe(0) типа ML5. А здесь по результатам лигандного обмена – мы видим, что новый лиганд вынес L+2X – мостиковый хлорид и два хлорида, из чего мы можем сделать вывод, что и способ связывания циклобутиена лучше описывается типом X2L, но это и есть 6-электронный дианион. Всё сошлось! Но оставило нас в состоянии крайней растерянности. Мы сказали, что 6-электронность циклобутадиена возможна, но обеспечивается через эффект back-donation. А здесь она сама собой образовалась через самый банальный лигандный обмен с уже готовым 6-электронным дианионом. И рутений в получившемся комплексе имеет степень окисления +2, а не ноль, а структурный тип циклобутадиенового комплекса тоже MX2L4, а не ML5. Так у таких же железных комплексов тогда тоже так, но только там то же самое достигается через back-donation и железо там тоже Fe(2+)? А вот как хотите, так и думайте. Одно точно можно сказать, рутений ничем принципиально от железа не отличается, и не должно быть такого, что кто-то нам скажет – вот у рутения так, а у железа сяк. Здесь дело не в металле, а в способе получения комплекса. (Код структуры COWZEU: K.Takanashi, V.Y.Lee, A.Sekiguchi, Organometallics 2009, 28, 1248)
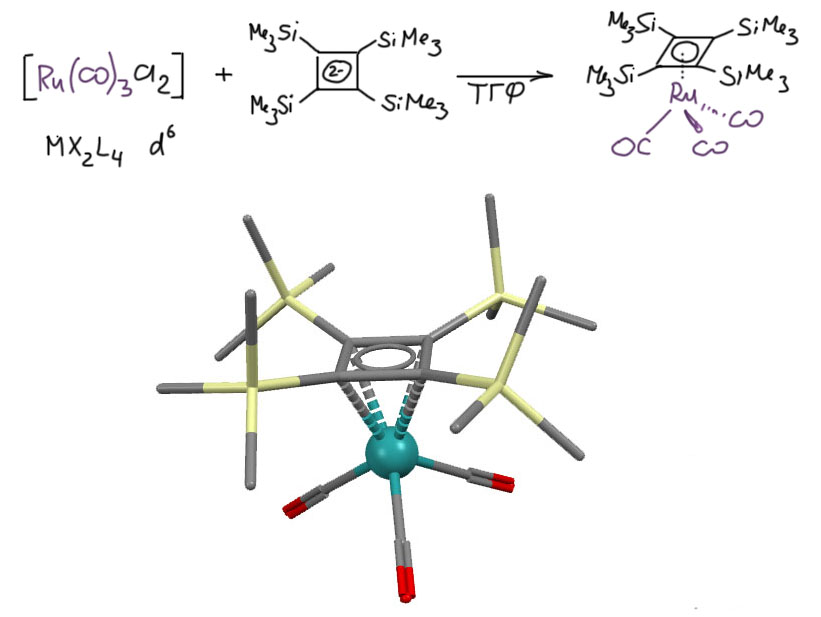
Этот ярчайший случай прямо вопит нам в уши – будьте внимательны с обсуждением структуры комплексов в тех случаях, когда возможно сильное проявление эффекта back-donation. В этих случаях всегда есть два способа посчитать степени окисления и конфигурацию металла, но, что самое приятное – общий счет валентных электронов от этого не зависит. В уже упомянутых комплексах что железа, что рутения мы имеем 18-электронные комплексы.
Координационная химия не выработала однозначных рекомендаций для таких случаев. А их, как мы уже видим, много. А увидим дальше, что их еще больше, и будут очень важные примеры таких соединений именно в катализе, а не только в рассматривании самих комплексов. По-моему, эта особенность низковалентных комплексов с лигандами, способными служить хорошими π-кислотами необычайно украшает эту науку, привносит в неё элементы хорошей головоломки. Сейчас мы займемся двумя другими циклическими системами, которые выдают еще более затейливую игру возможностей.
Ржавый гвоздь в гроб ароматичности
Конечно же это преувеличение и ароматичность еще всех сама похоронит, но всё же, некоторые проблемы есть.
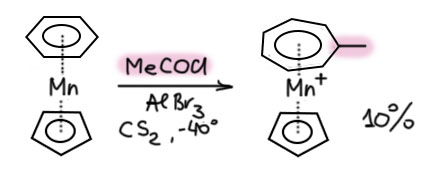
Комплексы переходных металлов испытывают наши догмы. Мы уже выяснили, что в виде лиганда нам до лампочки правила Хюккеля. Но окончательно распроститься с такими вещами очень трудно. Представим себе, что у нас есть комплекс какого-нибудь позднего переходного металла, у которого есть циклопентадиенильный лиганд – икона ароматической догмы, и еще циклобутадиеновый лиганд. То есть прям буквально кошмар Хюккеля. Такое бывает? Да, первый такой комплекс получил уже упомянутый Петтит, а вообще их известно немало. Это усточивые комплексы, вполне отвечающие образу сэндвичей, и оба многоугольника правильные – квадрат и пентагон. Первый комплекс Петтита был кобальтовым (Amiet, R. G., Pettit, R. J. Am. Chem. Soc. 1968, 90, 1059), и это было устойчивое диамагнитное соединение с идеальным спектром ЯМР из двух синглетов при 4.86 (5H) и 3.61 (4H) м.д. Петтит не стал церемониться и вдарил по этому комплексу точно так же как это делал Вудвард с ферроценом – хлористым ацетилом и хлористым алюминием. Чудес не бывает, по стабильности подойти близко к ферроцену трудно, почти все осмолилось, но с выходом 10% всё же получился продукт ацетилирования с отличным спектром ЯМР с сигналами при 4.87 (5H), 4.28 (2H), 3.85 (1H), 1.88 (3H).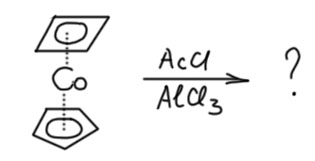
Разберитесь что получилось, Псчитайте электроны в комплексах. Объясните, почему получился именно этот продукт, почему выбрано именно это кольцо для электрофильного замещения. Предложите структуру для промежуточного комплекса и объясните как он устроен.
В зависимости от полноты ответа можно получить до 20 плюсиков.
Циклогептатриенил
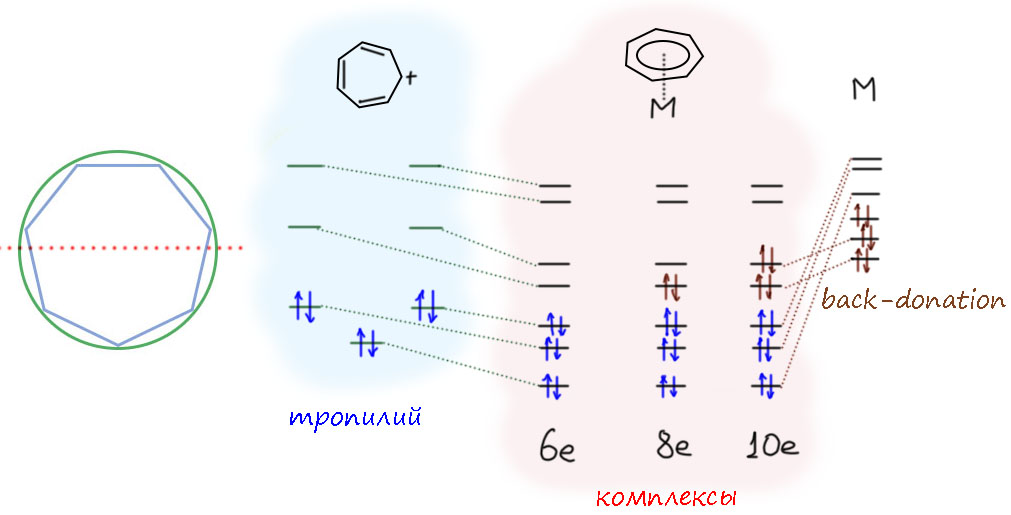
Циклогептатриенильный катион (тропилий) – еще одна классическая ароматическая система. Не удивительно, что комплекс с таким лигандом попытался получить уже сам Уилкинсон сразу после открытия ферроцена, но попытка “в лоб” к успеху не привела – вместо комплекса был просто получен продукт восстановительного сдваивания, циклогептатриенила. Задача непроста, ведь ароматический циклогептатриенил – это катион, хотя это довольно формальное обстоятельство, и из общих соображение не видно, почему эта система не может быть шестиэлектронным лигандом. Но только вот вопрос – а как он связан? Это очень непростой вопрос, а этот лиганд, который очень хорошо известен в комплексах с самыми разными металлами, пожалуй может быть назван самым загадочным в смысле анализа его связывания.
Сложный характер связывания проявился уже в самом первом исследовании Даубена и Хоннена (H. J. Dauben Jr, L. R. Honnen, J. Am. Chem. Soc. 1958, 80, 5570). Начинали с реакции циклогептатриена с карбонилом молибдена, получили вполне банальный комплекс триена с тригапто-связыванием, Mo(0), d6, ML6. Дальше от связанного циклогептатриена отрывали гидрид обычным способом – гидридным переносом на трифенилметильный катион. Состав комплекса при этом не изменяется, хотя лиганд уже другой гептагапто, но можно считать, что он по прежнему 6-электронный, комплекс имеет заряд 1+. Считаем, что это [ML6]+, но это плюс на лиганде. Следующая реакция очень простая, но она сразу ставит вопросы. Это просто лигандный обмен нейтрального карбонила на иодид-ион. Получается нейтральный комплекс, понятно почему – это формальная нейтрализация двух противоположных зарядов. Но какова степень окисления молибдена? Иод – точно X-лиганд, и вроде бы должен давать молибдену вкада +1 в степень окисления. Но Mo(1+) – экзотическая степень окисления для этого металла, и она должна создавать парамагнитность. Хорошо, посмотрим следующую реакцию, может быть она нам что-то прояснит: подействовали циклопентадиенилом, который входит обычным пентагапто-способом в сферу, этот лигандный обмен должен вытеснить 2L+X, и мы видим, что ушёл иодид, и сильно изменился способ связывания циклогептатриенила – он превратился в обычный аллильный лиганд, то есть L+X. Итого, последний комплекс имеет тип MX2L5, молибден в нём Mo(2+) – это вполне типичная степень окисления для этого металла. Итого в последнем комплексе d4 и 14 электронов от лигандов – это 18-электронный комплекс. Отыграем назад – скорее всего структурный тип при лигандном обмене не изменился. И тогда на циклогептатриенил приходится … X + 3L, а это значит, что и в нем молибден уже имеет степень окисления 2+, а циклогептатриенильный лиганд – 8-электронный!! Если бы мы только что не разобрали приключения циклобутадиена и не поняли, что смотреть на “антиароматический” счет электронов не нужно. И здесь мы имеем формально циклогептатриенильный анион. А это значит, что этот переход произошел уже в предыдущем комплексе. Молибден за счет back-donation отдал пару электронов циклогептатриенилу, сделал его лигандом типа XL3.
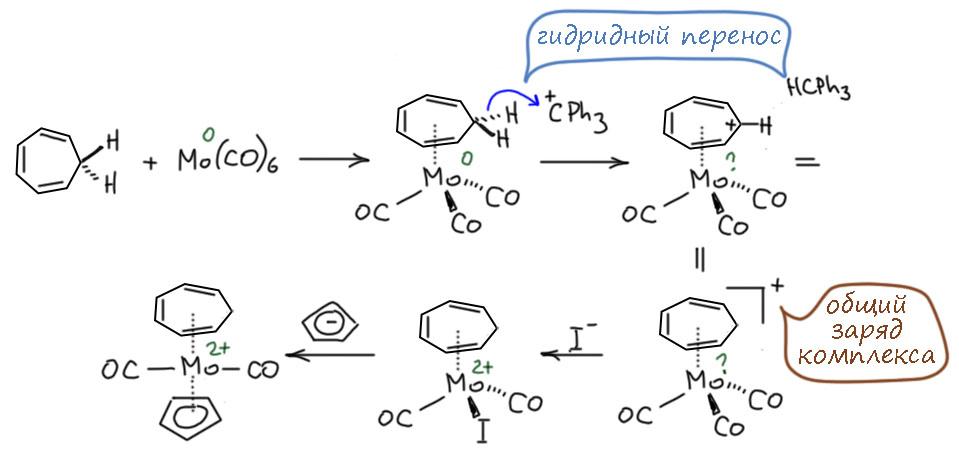
Ещё раз посмотрим на циклогептатриенильный лиганд. Вспомним молекулярные орбитали (их расположение, сами орбитали разрисовывать не будем, большой пользы в этом не будет) этой системы, которую кратко называют тропилием. Их легко рисуют с помощью круга Фроста. У самого тропилия большинство орбиталей (все кроме самой нижней) дважды вырождены. У тропилия шесть электронов на пи-МО. При связывание в комплекс всей системой целиком – гептагапто-способом – орбитали переходят в комплекс как-то комбинируясь с орбиталями металла, при этом вырождение снимается, поскольку понижается симметрия (почему это обязательно мы уже рассмотрели, обсуждая циклобутадиен). Если взаимодействие обеспечивается только координационными связями (металл – только кислота Льюиса, тропилий – только основание), то лиганд остается 6-электронным. Большой вопрос – существуют ли вообще примеры таких комплексов. Если металл может и имеет электроны для back-donation, то передача одной пары сделает лиганд 8-электронным. Это будет формально циклогепратитриенил-анион. В обычной химии такая система антиароматическая, если плоская, или неароматическая, если искажена. В комплексах никакой антиароматичности нет, в восьми электронах проблем нет, и лиганд такого типа будет связываться как XL3. Но это еще не все, есть и еще одна орбиталь для back-donation, и если у металла есть такая возможность, лиганд станет 10-электронным, и будет связываться по типу X3L2.
Трометцены
Циклогептатриенильный лиганд оказался очень интересным для сэндвичевых комплексов металлов с двумя параллельными циклами, сделанными по подобию ферроцена, но так, что один лиганд – циклопентадиенил, а второй – циклогептатриенил. Такие комплексы известны для многих металлов, у них очень интересная химия, и в самые последние годы их стали использовать для самых разных применений. Для красоты и понтов им придумали прикольные названия – тро-мет-цены. Тро – это циклогептатриенил, потому что катион такой называется тропилий. Увы, мы уже понимаем, что в комплексах этот лиганд нечасто бывает тропилием, а остальные производные так вроде бы называть нельзя – вот как назвать такой анион? Тропилий-анионом нельзя, ведь окончание -илий означает именно катион. А как? Да никак, только целиком, но для того чтобы дать три буквы -тро- в название годится, не будем брюзжать. Итак, берут конкретный металл и делают название. Титан – тротицен. Цирконий – троцирцен, гафний – трогафцен. Ванадий – тровацен. Почему-то для производных ниобия и тантала таких названий не применяют, хотя были бы вполне нормальными – трониоцен и тротанцен – даже красиво. Хром – трохроцен – хорошее слово, надо использовать для обозначения неприятных персонажей: этот трохроцен опять несет вздор… и т.п., впроцем трогафцен не хуже. Молибден и вольфрам почему-то тоже не используют, были бы тромоцен и тровольцен (по-английски случился бы тротанцен и коллизия с танталом – может поэтому не используют оба). Марганец – троманцен. На этом всё. Такие комплексы есть не для всех металлов. Посмотрим почему.
Циклопентадиенил это XL2. Циклогептатриенил возьмём для начала 10-электронным X3L2. В сумме 16 электронов. В степень окисления дают +4. Тротицен и вся четвертая группа тогда будут комплексами M(4+) с 16-ю электронами, это нормально, в 4-й группе полно 16-электронных комплексов. Пятая группа: берём M(4+) получим 17-электронные комплексы ванадия и других, для этой группы парамагнитные 17-электронные комплексы очень свойственны взять хоть ванадоцен, в котором V(0). Шестая группа: лучше всего, ведь для M(4+) будут 18-электронные комплексы, причем их же можно трактовать как комплексы с 8-электронным циклогептатриенилом, который представляем как XL3 – в степень окисления +2, сумма двух лигандов 14e. А как правильно? Как хотите, критериев однозначного определения степени окисления, хоть экспериментальных, хоть теоретических не придумано, хотя анализ электронной структуры, полученной хорошим расчетом какие-то намёки даёт. Но нам это нафиг не нужно. Но иногда определенность есть. Вернемся опять в 4-ю группу, к титану и компании. Если взять 8-электронный лиганд, то металл будет иметь степень окисления +2 и конфигурацию d2. Эти два электрона – это живая пара, одна. У титана таких комплексов воз и маленькая тележка и они всегда выглядят как такие карбены – два лиганда уголком, и в другую сторону кокетливо торчит пара на орбитали. Эта пара не позволяет кольцам быть параллельными. Но они параллельны. Следовательно там нет пары, металл имеет степень окисления +4, и по крайней мере для начала ряда (3-ю группу не берем) гипотеза о 10-электронном циклогептатриениле ближе к реальности. Для пятой группы этот аргумент уже не будет столь же бесспорным. С другой стороны, комплекс рутения скорее имеет 8-электронный лиганд и Ru(4+), потому что с 10-электронным лигандом степень окисления стала бы 6+, а это маловероятно.
Переходим в 7 группу. Если лиганд 10-электронный, то Mn(4+) с конфигурацией d3 даст 19 электронов. Если 8-электронный, то Mn(2+) и d5 и те же 19 электронов. Перелёт! Такой комплекс существует с зарядом +1, то есть Mn(5+) или Mn(3+) – троманцений. Ещё есть один комплекс в 8-й группе у рутения и это дикатион. Понятно, что в 9-й и 10-й группе потребовался бы трикатион или тетракатион и это перебор, степень окисления металла стала бы для поздних переходных металлов неприемлемо большой. Известные трометцены даны по статье большой группы австрийских и немецких исследователей: R. Basse, S. Vanicek, T. Höfer, H. Kopacka, K. Wurst, T. Müller, H.A. Schwartz, S. Olthof, L.A. Casper, M. Nau, R. F. Winter, M. Podewitz, B. Bildstein Organometallics 2021, 40, 2736 – то есть это очень свежие данные, и если у ряда будет продолжение, то в будущем.
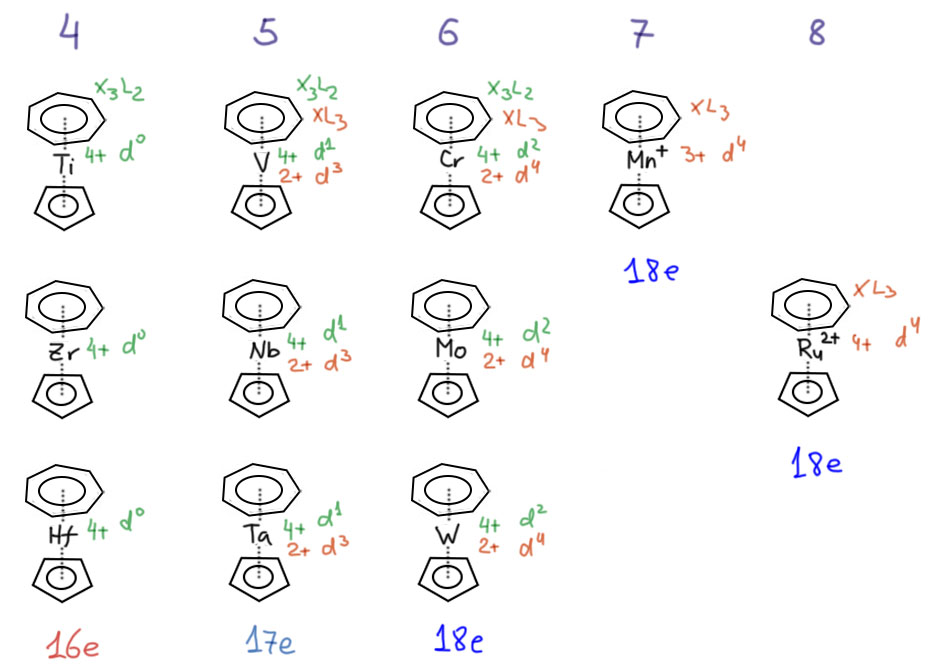
Видим, что комплексов много, что в основном они у ранних переходных металлов. Получается, что эти комплексы интересно дополняют обычные металлоцены, которые, конечно, перекрывают весь ряд кроме 11 группы, но для первых двух групп металлоцены бывают только гетеролептические со скособоченными циклопентадиенилами, а самые крутые металлоцены это все же 8-я группа и рядом.
У трометценов интересная и разнообразная химия. Многие из них устойчивы, легко получаются, вступают в разнообразные реакции с сохранением системы и отлично годятся как основа для построения более сложных комплексов. Подробнее обсуждать не будем, если они на понадобятся для какого-нибудь катализа, тогда и разберёмся. Но одну историческую вещь не могу не упомянуть. Самый первый раз комплекс такого типа был случайно получен тем самым Отто Фишером с помощью очень странной реакции. Фишер попробовал проацилировать комплекс марганца с бензолом и Cp. Очевидно, что ожидалось, что ацил пойдет нормально и чистенько в циклопентадиенильное кольцо. Но даже при том, что условия реакции были очень мягкие, все к черту осмолилось, но упорный Фишер смог из этого осадить с помощью обычного приема – в виде соли с большим противоионом – катионный комплекс, который оказался … троманцением. Ацетил встроился в бензол, Cp кольцо вообще осталось нетронутым.
Как произошло это внедрение карбонильного углерода можно только гадать, очевидно что это катионная перегруппировка. Мы даже, к сожалению, не можем сказать была ли атака по циклопентадиенилу, потому что такие продукты могли осмолиться. Но реакция во-первых, очень интересная, и во-вторых, она косвенно говрит о высокой стабильности трометценовой системы.
Циклооктатетраен
Следующий и последний возможный лиганд этого типа – циклооктатетраен. А девять? Ведь есть такой ароматический аннулен? Да, есть, в виде моноаниона, по аналогии с циклопентадиенильным анионом, но он уже не очень устойчив, цикл сильно напряжен, и хотя исключить возможность получения комплексов с таким кольцом нельзя, это скорее всего будет нечто с меньшей гаптностью, и не очень интересное. А вот циклооктатетраен – лиганд очень популярный и интересный.
Сам циклооктатетраен (COT) – отлично известная неплоская молекула, существующая в виде “ванны”, где все двойные связи имеют цис-конфигурации, и это довольно жесткая штука, в которой при обычных условиях нет существенных конформационных движений. При повышенных температурах подвижность появляется и ванна может выворачиваться, но все равно в другую ванну. Из общих соображений можно представить и кресло, в котором две связи транс, а две цис и может показаться, что это должно быть выгоднее. Но нет, это не так, и в общем даже понятно почему – такая структура должна иметь очень большие угловые напряжения. 8-членный цикл – первый по размеру, в котором может быть одна транс-двойная связь, но ей там не очень уютно, даже когда всё остальное – более-менее гибкая насыщенная цепь из 6 атомов. А когда остальные атомы тоже sp2 и свободы еще меньше, такая структура становится очень напряжённой.
Поэтому ЦОТ сильно отличается от всего, что было до этого. Исходный лиганд изначальное неплоский и больше напоминает диен, да и в реакциях проявляет свойства диена. Циклооктатетраен становится плоским, только если получает ещё электроны. Дианион циклооктатетраена хорошо известен, и это правильный восьмиугольник, который считается примером 10-электронной ароматической системы, [8]-аннулена. Соль дикатиона с катионами калия легко получить Может ли такой лиганд быть 10-электронным лигандом? Если может, то как это достигается. Собственно есть два очевидных пути:
- лиганд связывется как нейтральный L4 и в координационной сфере получает два электрона через механизм back-donation, и мы формально учитываем это, изменяя степень окисления металла на +2, а способ связывания лиганда на L3X2.
- лиганд связывается как дианионный, металл сохраняет степень окисления, а способ связывания сразу считается L3X2
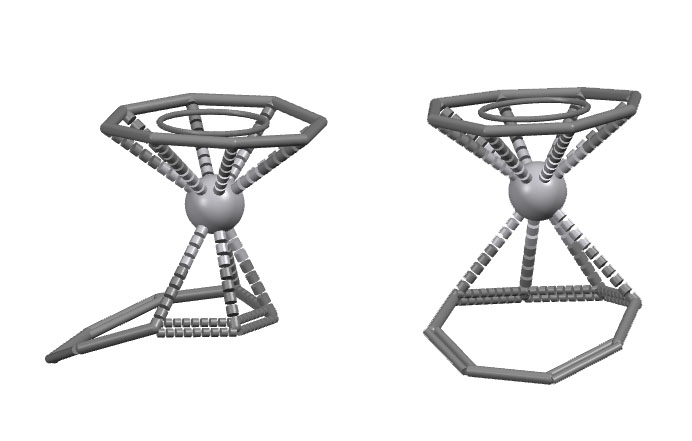
Если при этом мы получаем десятиэлектронный η8-лиганд, мы должны понять, а есть у нас в валентной оболочке металла орбитали, способные опрходовать пять пар электронов. Нам нужны 5 атомных орбиталей, обладающие возможностью перекрывания в направлении одной оси координат, для простоты мы всегда выбираем ось z. Да, есть и сохраняя стандартные обозначения это s, pz, dz2, dxz, dxy – как раз пять штук. Отсюда следует, что такой лиганд мы можем ожидать увидеть у ранних переходных металлов в высоких степенях окисления и конфигурацией не более d4, но скорее d0 и d2. Запомним это, но сначала подумаем про возможность образования сэндвичевых комплексов с двумя такими лигандами. Это большой соблазн – получить такие аналоги обычных ценов, с такими большими ломтями на сэндвиче, и понятно, что чей-то рот сильно бы обрадовался такому куску. Впервые комплекс с двумя такими лигандами получил крупнейший немецкий металлоорганик Гюнтер Вильке в 1966 году для титана – в статье (H. Breil, G. Wilke, Angew. Chem. Int. Ed. 1966, 5, 898) – это был бы 20-электронный комплекс титана(4+). Поскольку Вильке простаком никогда не был, он не стал заявлять, что получил такое чудо до надежного установления структуры, но этого сделать не получилось, кажется, до сих пор – комплекс оказался сильно разупорядоченным, а это само по себе скорее говорит против высокосимметричной структуры.
Такие комплексы получить тем не менее удалось, но для этого понадобились f-элементы, причем сначала такие, которые могут иметь степень окисления 4+, а это среди лантанидов церий, но среди актинидов многие. Я уже давал торжественную клятву не лезть в f-элементы в этом курсе. Вообще, это довольно странные элементы. Казалось бы, это должен быть еще один шаг в сторону усложнения от переходных металлов, и мы должны бы увидеть совсем чудесные комплексы и реакции. Но f-оболочка не совсем валентная, она скорее просто экранирует настоящую и очень скромную валентную оболочку фактически типичного элемента 3-й группы, и вступает в дело у лантанидов вообще только тогда когда на ней 1 электрон или половина (семь) плюс-минус один электрон. Дело в том, что 4f-оболочка довольно компактна и она просто утоплена в нагромождении невалентных, внутренних орбиталей, особенно весьма диффузной 5p-оболочки, и эти никудышние внутренние электроны просто отталкивают всех желающих зацепиться за f-орбитали. У актинидов 5f-оболочка намного более протяженна и без труда, по крайней мере у первых элементов этого семейства, выбирается из-под опеки остовных оболочек. У f-элементов есть тем не менее два с половиной преимущества в отношении данной задачи – они скорее примыкают к ранним переходным металлам, и имеют приличные размеры электронных оболочек, а последнее особенно важно для того, чтобы нахлобучить себе на голову такой большой таз как восьмиугольник COT, и усесться на второй такой таз. А также, по крайней мере интуитивно, не обязаны трястись при виде 18-электронного правила и могут себе позволить слегка его бортануть, а это значит, что f-оболочка всё же участвует в связывании, и тогда хватает валентных возможностей на оба блина, хотя там весьма значительна ионность связей с лигандами. Вот и первый цен с двумя октагонами получили для урана, и сделал это в 1969 году универсальный американский химик Эндрю Стрейтвизер (A. Streitwieser, U. Müller-Westerhoff, J. Am. Chem. Soc. 1968, 90, 7364), так и назвав полученное соединение ураноценом. Структуру удалось получить только для нескольких замещённых ураноценов, например, вот такого (код структуры BIJXIB – A.Zalkin, D.H.Templeton, W.D.Luke, A.Streitwieser, Organometallics 1982, 1, 618). Просто посмотрим на два одинаковых блина, и убедимся, что это и правда настоящий сэндвичевый комплекс, причем, на довольно близком расстоянии от металла.
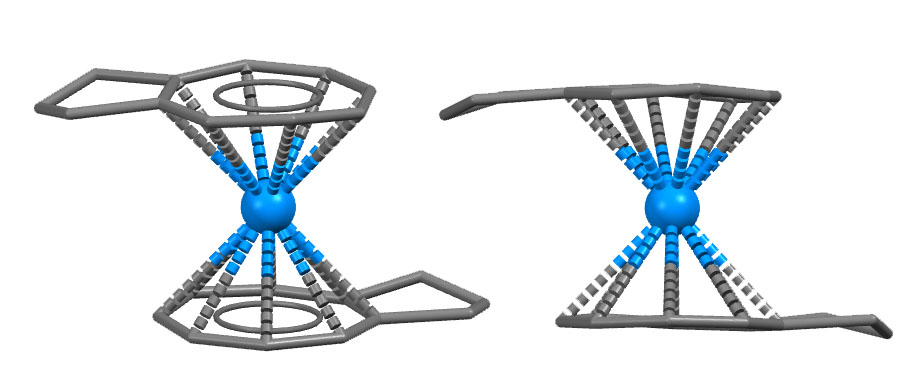
Видно, что блины немного кривые. Что-то там слегка не так, впрочем, это может быть следствием некоторой разупорядоченности молекул в кристалле. Но не так давно научились делать и комплексы лантанидов с двумя ЦОТ-блинами, причем металл в этих комплексах имет степень окисления +3, а общий заряд комплекса тогда становится -1. 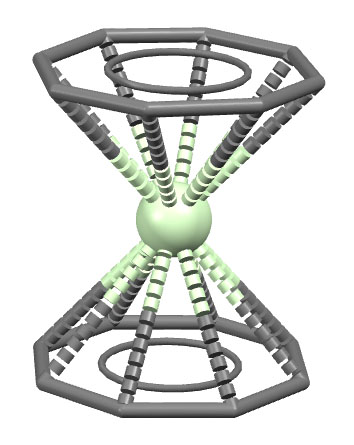
Вот пример такого комплекса, можно сказать, празеодимоцен (код структуры DEHQEP: C.T.Palumbo, M.E.Fieser, J.W.Ziller, W.J.Evans, Organometallics (2017), doi:10.1021/acs.organomet.7b00498). И в этих комплексах геометрия идеальна. Как они устроены? Скорее всего, в значительной степени за счет ионной связи, хотя формально это 20-электронные комплексы, но запихнуть все 20 электронов на орбитали металла вряд ли возможно.Интересно,что в комплексе лантанида, празеодима, расстояние до колец от металла даже чуть больше, чем в ураноценах. Но ничего необычного в этом нет. Это только обыденное сознание подсказывает, что уран по размеру намного больше любого лантанида, но нет, не больше, а в виде иона U(4+) даже немного меньше, чем Pr(3+), если судить по ионным радиусам, но раз мы договорились, что связывание в таких комплексах в значительной степени ионное, то какими ещё радиусами нам пользоваться.
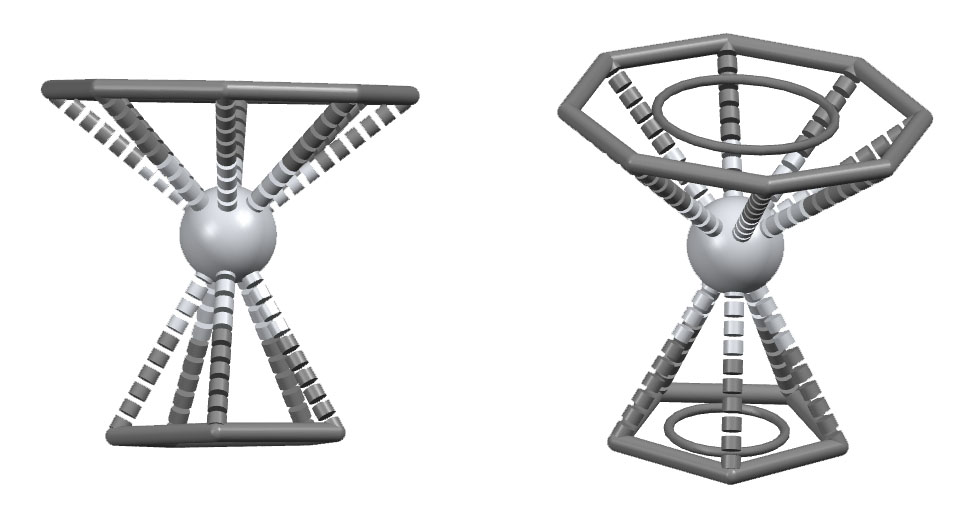
Всё, бросим f-элементы, они забавные, но какие-то тоскливые, и не очень хорошо у них работает наша схема с подсчетом валентных электронов и преобладанием ковалентных связей, и вернёмся к нашим привычным переходным металлам. Настоящие переходные металлы не потянут два полноценных ЦОТ-блина, и бис-комплексы, известные для ванадия и титана, имеют другую структуру. Вот, ванадиевый комплекс (структура SUBCOH, D.Gourier, E.Samuel, B.Bachmann, F.Hahn, J.Heck, Inorg.Chem. 1992, 31, 86). Сослепу можно даже подумать, что он обычный, особовенно если не повезёт с углом зрения. Но, если рассмотреть получше, видно, что если одно колько нормальное, плоское, то второе какое-то скособоченное, и если рассмотреть, то оказывается, второе кольцо цепляется как диен η4-способом. Возникает вопрос, что это за второе кольцо. И что за ванадий. Комплекс получают из трёхвалентного ванадия реакцией со смесью дианиона COT и свободного COT, а в комплексе можно предположить V(4+). В этом месте становится немного не по себе. Если кто-то еще не забыл, чему учили в школе, то превращение V(3+) в V(4+) это окисление. А где окислитель? Это дианион COT окислитель!!! Нет, это невозможно. Тут мы замечаем в реакционной смеси свободный COT, мы сразу не поняли зачем это, но оказывается это и есть окислитель. Но тут опять неувязочка – если этот COT принимает два электрона, то ванадий должен был бы быть пятивалентным, но тогда у него должен быть еще один лиганд, а комплекс должен был бы быть диамагнитным (d0 на металле и дианион на обоих лигандах). Но комплекс точно парамагнитный, поэтому остается только принять, что это комплекс V(4+) с двумя дианионами, и более того, внимательно рассмотрев методику, чего мы обычно не делаем, мы видим вот такую стехиометрию – добавляют именно полэквивалента свободного COT, чтобы одного электрона от ванадия хватило бы на образование недостающего полэквивалента дианиона. В обшем, это тот случай, когда как устроен комплекс только по структуре не поймешь, пока не посмотришь еще и как он получился. Итого V(4+), конфигурация d1 и одно кольцо 10-электронное (X3L2) и второе связано как диен (ещё L2) – но это будет 15-электронный комплекс. Недобор. Хотя и исключить такое трудно, но есть и второй способ – учесть второе кольцо как 6-электронный лиганд, как LX2. В самой статье по этому поводу написана сущая чущь – сказано, что это типа, как будто циклобутадиенильный дианион, типа тоже ароматический. Простим это авторам, ибо не ведают, что городят. Им просто хочется получить 17 электронов. И мне даже не очень нравится идея про LX2, потому что диен с такой координацией обычно пристыковывается к металлу двумя крайними углеродами, образуя такой металлацикл, а тут чисто плоская координация как в обычных тетрагаптокомплексах диенов. В общем, мы имеем здесь комплекс с весьма двусмысленной схемой связывания и не до конца понятным способом присоединения второго кольца.
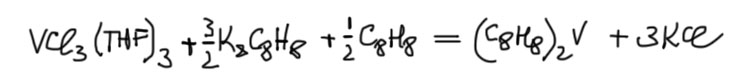
В общем смонтировать два восьмиугольных блина на нормальном переходном металле толком не вышло. Зато есть немало комплексов, где только один COT, а второй лиганд, например, обычный Cp. И тогда COT мохет быть идеально плоским, как например, вот в этом титановом сэндвиче (Van Oven, H. O.; de Liefde Meijer, H. J. J. Organomet. Chem. 1969, 19, 373.), полученном реакцией Cp2TiCl2 или CpTiCl3 с дикалиевой солью СОТ, это комплекс с неспаренным электроном, чувcтвительный к воздуху, но термически стойкий (код структуры CPCOTI: Kroon, P.A.; Helmholdt,R. B. J. Organomet.Chem. 1970, 25, 461). Если учитываем COT как L3X2, то весь комплекс имеет структурный тип TiL5X3 и 17-электронный счет, что вполне согласуется с парамагнитностью. Очень интересно сравнить этот комплекс с только что рассмотренным тротиценом, и убедиться, что все работает, и поэтому ясно почему этот комплекс парамагнитный, а тот диамагнитный. Мне ясно, а вам? Интересно заметить, что из двух блинов верхний вроде бы нахлобучен поглубже, он ближе к атому металла. Но это только потому что он шире, а расстояния Ti-C для обоих блинов одинаковые с точностью до второго знака. То есть как бы не был связан лиганд, всё равно двухцентровые взаимодействия в химии важнее всего.
А вот у поздних переходных металлов нет возможности удерживать этот лиганд в его полной конфигурации, и он перестает изображать из себя что-то ароматическое и связывается, например, как хелат через две двойные связи в комплексе кобальта (COT)CoCp (код структуры NUTHEP: H.Wadepohl, R.Merkel, H.Pritzkow, Acta Crystallogr.,Sect.C:Cryst.Struct.Commun. 1998, 54, 1095), и это 18-электронный комплекс CoXL4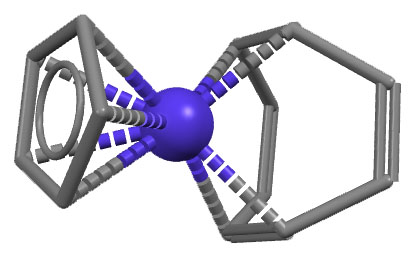
Довольно часто можно встретить комплексы, в которых плоский, а значит, скоре всего, 10-электронный ЦОТ связан с металлом не сэндвичевым способом, а меньшей гаптностью. Значит ли это, что back-donation может помочь впрыснуть два электрона на кольцо не сразу “в серединку”, а сбоку, через такую калиточку одинарной двойной связи? Такое бывает? Посмотрим, какая занятная химия получилась у голландских металлооргаников Клазинги, Тойбена и сотрудников (код структуры COCPTA: F.van Bolhuis, A.H.Klazinga, J.H.Teuben, J.Organomet.Chem. 1981, 206, 185). Исходный комплекс тантала(3+) имеет гидрид и η2-олефин и это координационно-насыщенный комплекс с 18е. Этот комплекс реагирует с ЦОТ. Как? Он же насыщенный. Скорее всего у комплекса есть равновесие с алкильным комплексом (гидрид тантала присоединяется к двойной связи), причем степень окисления металла не изменяется, а счет электронов уменьшается до 16. Это равновесие, скорее всего, смещено в сторону исходного комплекса (обратная реакция называется β-гидридным элиминированием, и мы часто с ней будем встречаться), и реакция именно этого комплекса с ЦОТ дает дигапто-комплекс, в котором происходит смещение электронов на лиганд так, что тантал фактически переходит в Ta(5+) а новый лиганд получает еще два электрона до плоского дианиона. Но в координационной сфере металла места осталось только для двухэлектронного лиганда, поэтому циклооктатетраен цепляется дигапто-способом. Интересно, что лиганд хоть и плоский, но довольно сильно искаженный, с разными длинами связей, такое впечатление, что они альтернируют, то есть чередуются по длине, как у неароматического полиена. Авторы и их последователи называют такой лиганд полуароматическим, опираясь на хорошо известный факт из химии циклооктатетраена: 10-электронный дианион у этой молекулы плоский правильный восьмиугольник, а 9-электронный анион-радикал тоже плоский, но с чередованием связей длинная-короткая. То есть в этом комплексе мы очевидно видим очередное следствие того, что эффект back-donation никому не обязан развиваться на полную катушку. С другой стороны, нам очень нравится идея, что за счет этого эффекта ранний переходный металл достигает максимальной степени окисления и опустошает свою d-оболочку, сгружая ее целиком в лиганды. И так как у этого комплекса вроде бы нет никаких признаков парамагнитности (впрочем, это ни о чем точно не говорит, так как Ta(4+) и анион-радикальная форма лиганда могли спарить свои спины, образовав синглетную форму с двумя неспаренными электронами на лиганде и металле, но с противоположными спинами), мы можем высказать другую гипотезу – дигапто-связывание дианионной 10-электронной формы рассиммметризует ее и делает связи по кольцу неравноценными. А уж к этому комическому термину “полуароматический” мы отнесёмся снисходительно, ни на минуту не забывая, что Ян Тойбен это не лох какой-нибудь, а один из ведущих металлооргаников и исследователей полимеризации Циглера-Натты. Но забавная штука получилась – такое походное опахало с ручкой – крутите ручку и цикл ЦОТ машет туда-сюда, создавая приятный ветерок. Хитры голландцы и мастеровиты, это еще знатный путешественник П.А.Романов замечал.
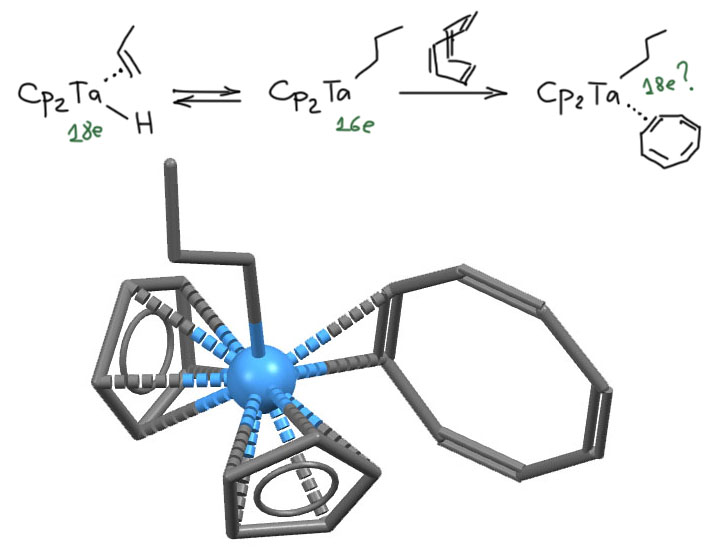
Поиграем в блинчики?
Итак, у нас есть аннуленовые лиганды от 3 до 8 атомов. Такие лиганды могут образовывать комплексы сэндвичевого типа – металл между двумя блинами. И никаких других лигандов. Задача – прикинуть, с металлами каких групп можно ожидать образования самых устойчивых комплексов этого типа для всех вариантов комбинации циклов (3-3, 3-4, … 4-5, 4-6…. 5-5, 5-6… 6-6, 6-7 … 7-7, 7-8, 8-8). Каждый тип комплексов обоснуйте, прикинув степени оуисления металлов, конфигурацию металла.
За полный ответ можно получить до 30 плюсиков.
Разберитесь в структурах комплексов, посчитайте электроны, и прочие характеристики комплексов. Если подсчёт неоднозначен, дайте возможные варианты. За каждую структуру можно получить до 4 плюсиков.
 (t-BuPh2C3)Fe(CO)2(NO)
(t-BuPh2C3)Fe(CO)2(NO)
Hughes R. P., Lambert J. M. J., Whitman D. W., Hubbard J.L., Henry W. P., Rheingold A. L. Organometallics 1986, 5, 789
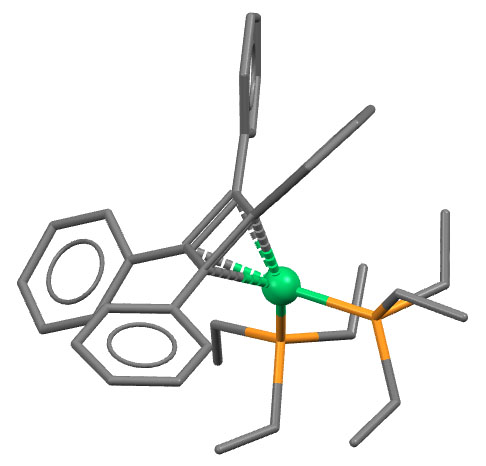
(Ph4C4)Ni(PEt3)2
J.J.Eisch, A.M.Piotrowski, A.A.Aradi, C.Kruger, M.J.Romao, Z.Naturforsch.,B:Chem.Sci. 1985, 40, 624
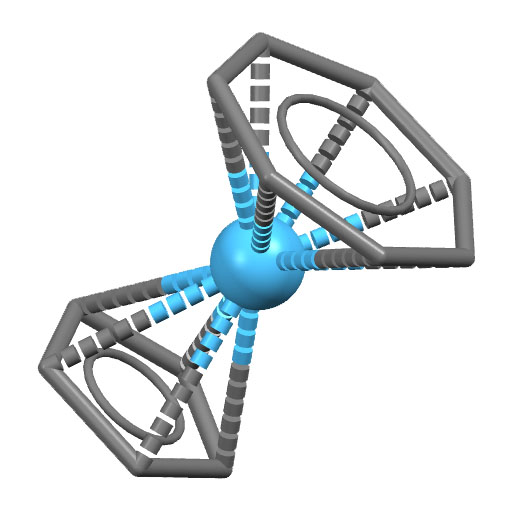
Трогафцен
S.Buschel, T.Bannenberg, C.G.Hrib, A.Glockner, P.G.Jones, M.Tamm, J.Organomet.Chem. 2009, 694, 1244, doi:10.1016/j.jorganchem.2009.01.055
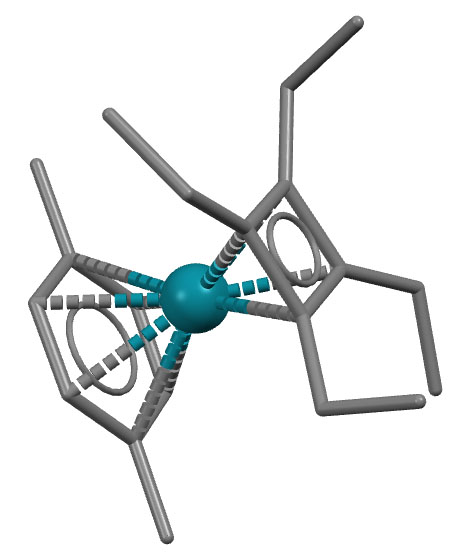
[(Et4C4)Rh(p-xylene)]+
D.S.Perekalin, N.V.Shvydky, Y.V.Nelyubina, A.R.Kudinov, Chem.-Eur.J. 2015, 21, 16344, doi:10.1002/chem.201503270
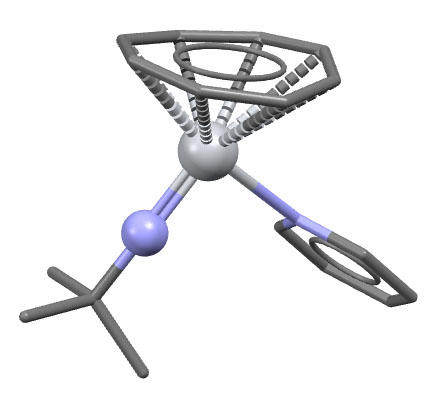
(cot)Ti(py)NtBu
S.C.Dunn, N.Hazari, A.R.Cowley, J.C.Green, P.Mountford, Organometallics 2006, 25, 1755
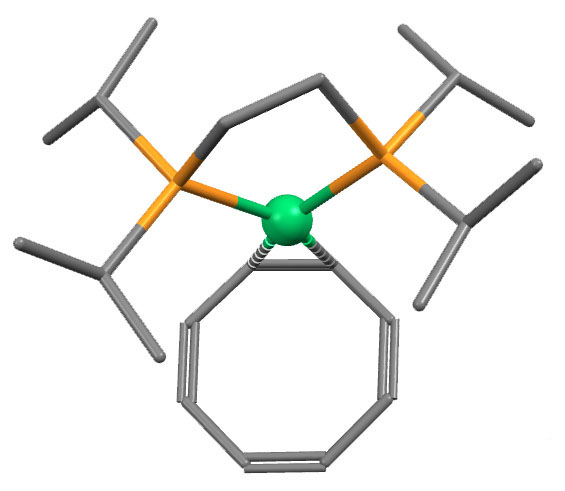
(COT)Ni(dipppe)
I.Bach, K.-R.Porschke, B.Proft, R.Goddard, C.Kopiske, C.Kruger, A.Rufinska, K.Seevogel, J.Am.Chem.Soc. (1997), 119, 3773, doi:10.1021/ja964210g
забыл металл шариком выделить, это зеленый никель
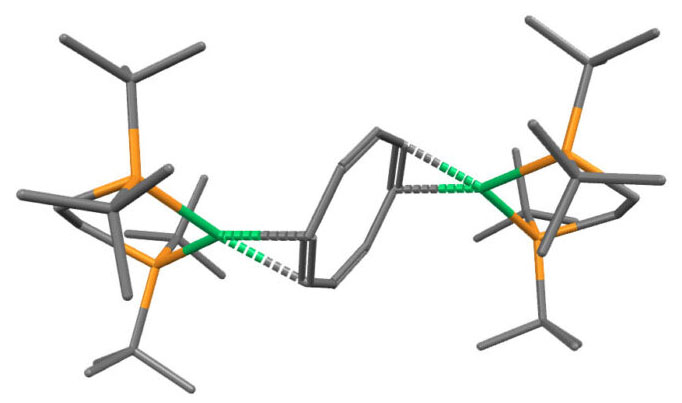
(dtbppe)Ni(COT)Ni(dtbppe)
I.Bach, K.-R.Porschke, B.Proft, R.Goddard, C.Kopiske, C.Kruger, A.Rufinska, K.Seevogel, J.Am.Chem.Soc. (1997), 119, 3773, doi:10.1021/ja964210g
забыл металл шариком выделить, это зеленый никель
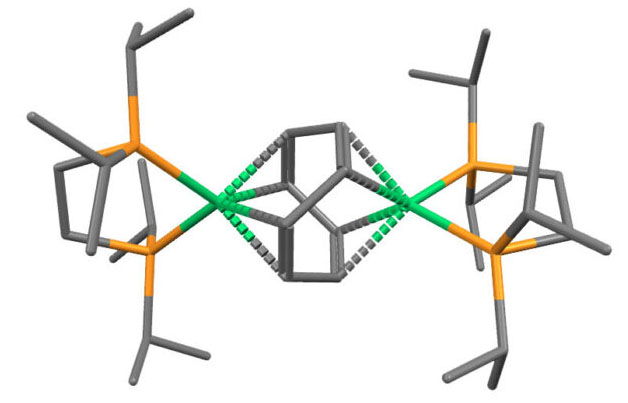
(dipppe)Ni(COT)Ni(dipppe)
I.Bach, K.-R.Porschke, B.Proft, R.Goddard, C.Kopiske, C.Kruger, A.Rufinska, K.Seevogel, J.Am.Chem.Soc. (1997), 119, 3773, doi:10.1021/ja964210g