
Аллильные комплексы палладия
Самый простой η3-аллильный комплекс палладия легко получается окислительным присоединением Pd(0) к аллилхлориду, очень стабилен, и очень часто используется как пред-катализатор в различных реакциях. Обычный синтез этого комплекса заключается в действии подходящего восстановителя на соль палладия и аллилхлорид, например, при пропускании CO в водно-метанольный раствор аллилхлорида и тетрахлоропалладата натрия. Оксид углерода очень легко взаимодействует с Pd(2+) с образованием карбонильного комплекса, который реагирует с нуклеофилом (воды или метанола достаточно) с образование Pd(0) и CO2. Как устроено взаимодействие металла и CO мы скоро изучим подробнее, поэтому пока просто схема реакции без подробностей:
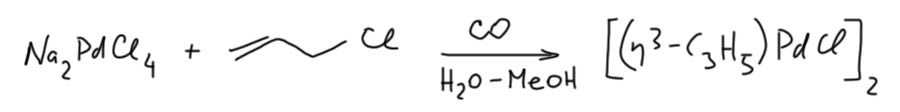 Он представляет собой димер с мостиковыми атомами хлора.
Он представляет собой димер с мостиковыми атомами хлора.
С другими аллильными комплексами проще взять сразу комплекс Pd(0) и аллильное галогенпроизводное и получить комплекс обычными реакциями замещения лигандов.
Вообще, палладий так любит η3–аллильные комплексы, что образует их и множеством других способов, например, при взаимодействии олефинов и диенов с солями Pd(2+). Из-за этого эти комплексы постоянно путаются под ногами там, где их никто не ждёт, и портят задуманные красивые реакции. Диены особенно легко их образуют, из-за чего, особенно в ранней химии, с диенами не могли сделать ничего толкового – всегда получались какие-то странные смеси продуктов. Современная химия, сильно насобачившаяся усмирять нежелательные реакции правильно подобранными лигандами, уже научилась работать и с диенами, и с другими капризными непредельными соединениями. Пока это оставим, но когда-нибудь обязательно обсудим.
Аллильное замещение
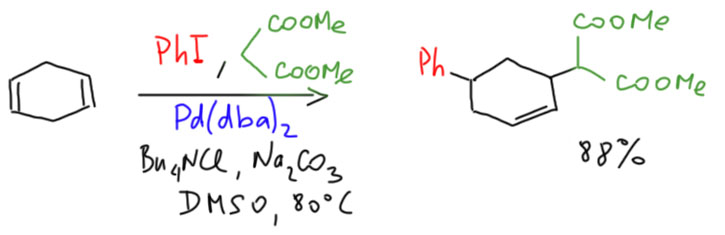
Образование η3-аллильного комплекса при окислительном присоединении Pd(0) к субстратам аллильного типа происходит настолько легко, что в этой реакции участвуют соединения с такими группами, которые почти никогда уходящими не являются ни в обычной органической химии, ни в реакциях окислительного присоединения с образованием σ-комплексов палладия, с которыми мы встречаемся, например, в реакциях кросс-сочетания. Первые патенты на эти реакции в начале 1970-х включали широчайший набор аллильных соединений, включая спирты, простые и сложные эфиры, даже амины. Патенты всегда пишут так, чтобы на всякий случай захватить поляну пошире, но в этом случае это не очень большое преувеличение, если не всё, то очень многое из этого списка действительно обладает способностью реагировать. Наиболее часто в этой химии встречаются сложные эфиры, как самые обычные типа ацетатов и бензоатов, так и чуть более редкие – карбонаты. Карбонаты – это эфиры угольной кислоты. Здесь речь идёт не о симметричных карбонатах (диаллилкарбонат – вполне банальное вещество, и в этой химии, естественно тоже реагирует, но мы потеряем один из аллилов, – невелика потеря, но в современной химии не принято разбрасываться органическими остатками), а о несимметричных, которые в изобилии встречаются в защите гидроксильных групп, типа Boc, Cbz, Fmoc и т.п.
Если в реакции участвует уходящая группа с кислородом, мы получим не только η3-аллильный комплекс, но и очень интересный побочный эффект, с которым мы не сталкивались в окислительном присоединении в кросс-сочетании. Дело в том, что палладий не очень любит кислородных лигандов. И если это сложный эфир, и уходящая группа ацетат или другой карбоксилат, ничего особенного мы не увидим – ацетат вполне работает как монодентатный или мостиковый лиганд:
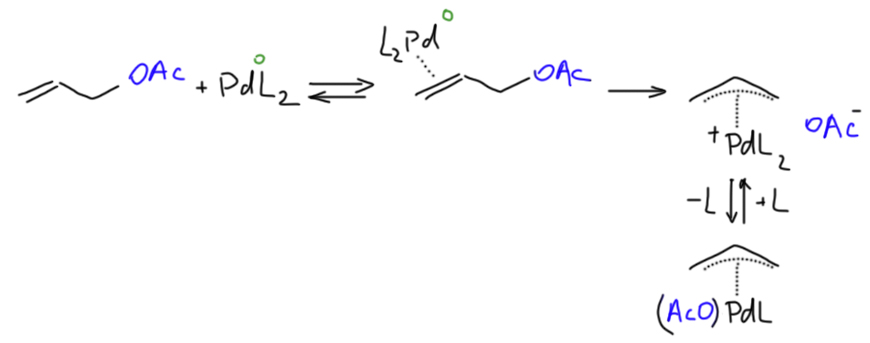
То в случае карбоната (возьмём для примера Boc-эфир аллилового спирта) образующийся анион монокарбоната неустойчив и разлагается с выделением CO2 и алкоксид-аниона. Последний совсем не нравится палладию в качестве лиганда – палладий предпочитает более мягкие лиганды. Вопрос – куда его девать? Это ведь весьма сильное основание.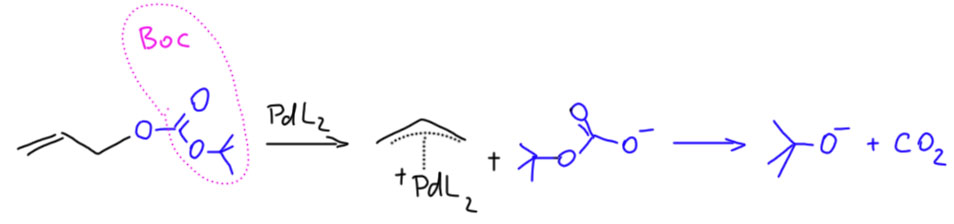
Цудзи с сотрудниками придумали, куда его девать – ему можно поручить отрывать протон от CH-кислоты, чтобы прямо на месте сделать карбанион (например, енолят), который и станет нуклеофилом в аллильном замещении (J. Org. Chem., 1985, 50(9), 1523). В результате получился очень мягкий метод аллилирования CH-кислот, самых разных. Понятно, что будут ограничения по pK – алкоксиды всё же не самые сильные основания. Простые енолизуемые кетоны, например, циклогексанон так не взять (чем можно взять циклогексанон Цудзи тоже придумал, тут где-то ниже есть пример такой реакции). Но то, что мы называем более кислыми CH-кислотами (малоновый эфир, кетоэфиры, нитроалканы и т.п.) реагируют чисто и мягко, например: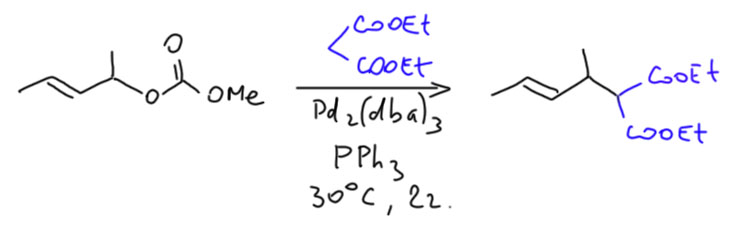
Вопрос. Винильные эпоксиды в аллильном замещении.
Из обычной органической химии мы отлично знаем, как раскрываются нуклеофилами эпоксиды. Довольно интересным объектом являются моноэпоксиды сопряжённых диенов, они очень легко получаются эпоксидированием диенов и представляют собой реагенты с большим потенциалом использования в синтезе. Но ещё больше этот потенциал раскроется при использовании реакций с участием комплексов переходных металлов.
Возьмём, например, моноэпоксид циклогексадиена-1,3. И раскроем его нуклеофилом, например, тем же енолятом малонового эфира. Получится продукт (кстати, почему именно этот?).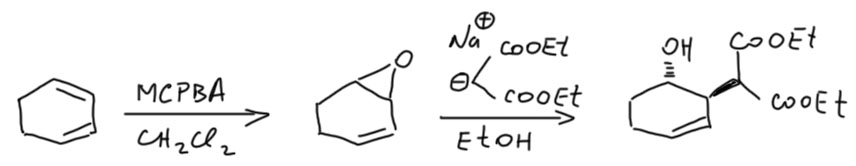
В таких эпоксидах есть аллильная система, поэтому можно попробовать его в реакции Цудзи-Троста. Они реагируют очень хорошо и прямо с малоновым эфиром, а не с готовым анионом. B получается при этом хоть и изомерный, но совсем другой продукт.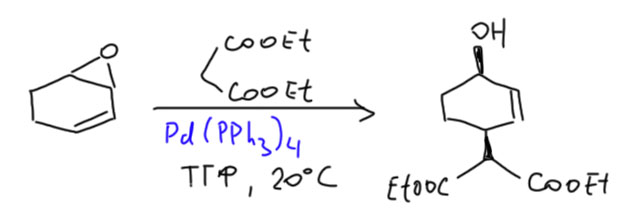
Как видим в случае винилэпоксидов (а это вполне общая реакция для моноэпоксидов сопряжённых диенов) палладий-катализируемое аллильное замещение и обычное аллильное замещение дают разные продукты. Попробуйте объяснить, как идёт палладий-катализируемая реакция со всеми её особенностями (возможность использования самой CH-кислоты, а не ее сопряжённого основания) и стереохимией.
B. M. Trost, G. A. Molander J. Am. Chem. Soc. 1981, 103, 5969
(до 25 баллов)
Стереохимия аллильного замещения
Важнейшей особенностью аллильного замещения, которое делает эту реакцию такой популярной в органическом синтезе является стереохимия. Но это очень противоречивая особенность – она и есть, и одновременно её нет. Нужно очень постараться, чтобы она проявилась – это не происходит, как в классической реакции SN2, автоматически. Попробуем разобраться, что в этой реакции такое происходит. Если посмотреть на то, как образуются аллильные комплексы, и как они реагируют с нуклеофилами, вроде бы всё очень понятно и красиво.
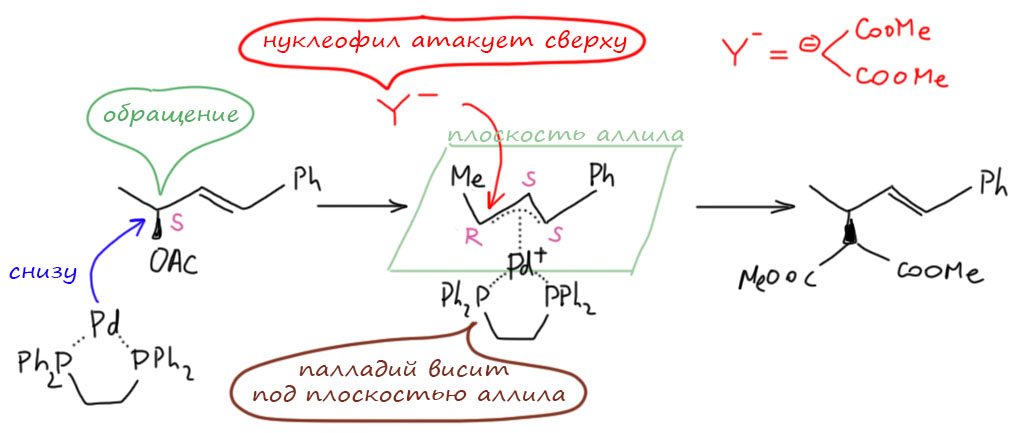
Окислительное присоединение и образование аллильного комплекса происходят стереохимически чисто с обращением конфигурации на том атоме, где была уходящая группа. Вся остальная часть аллильной системы фиксируется в той конфигурации, в которой она была в исходном аллильном субстрате. Впервые это экспериментально показал Тамио Хаяси с сотрудниками (T.Hayashi, T.Hagihara, M.Konishi, M.Kumada J. Am. Chem. Sос. 1983, 105, 7768). Взяв оптически активный аллильный ацетат они сделали реакцию с хелатным комплексом Pd(0) и получили оптически активный аллильный комплекс. Если расположить исходный ацетат так, чтобы ацетат торчал вперёд вверх, то палладий при образовании комплекса подходит снизу плоскости аллила, осуществляя обращение конфигурации на атоме с уходящей группой. Обратите внимание на R/S-разметку в комплексе: она сделана по обычным правилам, учитывая, что палладиевая часть принимается за заместитель максимального старшинства. Смотрите на комплекс сверху над аллилом, чтобы палладий висел снизу с другой стороны. На каждом атоме углерода будет ещё по водороду – это самая младшая группа. Дальше люди с развитым пространственным воображением на глаз определят конфигурации, а все остальные сделают очень простую вещь – продолжая смотреть сверху, мысленно меняем на каждом углероде водород и палладий – тогда у нас под взглядом будет ровно то, что требует правило – самый младший от вас – определяем убывание старшинства от палладия в каждой тройке прямо у вас под носом и определяем букву – настоящая конфигурация будет противоположной. Вот, на первом прохиральном атоме тройка – палладий, остальная часть аллила, метил – убывание по стрелке S, значит, конфигурация R, записываем. На втором атоме – палладий на месте водорода, часть с фенилом, часть с метилом – убывание по стрелке R, значит, S, записываем. На третьем атоме всё так же, фенил старше остальной части аллила. Обратите внимание, что для хиральности необходимо, чтобы на первом и третьем атоме аллильной системы были разные заместители (или хотя бы разные конфигурации, но это трудно сделать, поэтому оставим). Если бы они были одинаковые, мы получили бы типичную мезо-форму, правда, только если палладий со своим лигандом не разрушают плоскость симметрии, проходящую перпендикулярно плоскости аллила через второй атом.
Итак, комплекс – хиральная молекула. Продукт реакции оптически активного аллилацетата с нуклеофилом, например, с анионом малоната, любимой игрушкой в этой химии, тоже происходит стереохимически чисто – сверху от плоскости аллила. То есть если рассматривать палладиевую часть как заместитель, происходит обращение конфигурации – палладий фактически является уходящей группой. То есть по пути аллильного замещения мы имеем два обращения – при образовании комплекса и при атаке нуклеофила на комплекс. Как мы помним из обычной органической химии, два обращения = сохранению. Вот это мы и видим. Красота неземная. Хаяси не зря старался.
 Теперь спустимся с небес на землю и подумаем, а что так мучался этот Хаяси, нехилый, между прочим, учёный, очень много сделавший в этой области, если тут так всё чисто и красиво. Всё таки 1980-е годы – это вам не 1896 год, когда Павел Иванович Вальден открыл фундаментальное явление вальденовского обращения – техника эксперимента сто лет спустя сделала измерения оптической активности, да даже и абсолютной конфигурации, вполне рутинной работой для хорошего диплома, тем более в Японии.
Теперь спустимся с небес на землю и подумаем, а что так мучался этот Хаяси, нехилый, между прочим, учёный, очень много сделавший в этой области, если тут так всё чисто и красиво. Всё таки 1980-е годы – это вам не 1896 год, когда Павел Иванович Вальден открыл фундаментальное явление вальденовского обращения – техника эксперимента сто лет спустя сделала измерения оптической активности, да даже и абсолютной конфигурации, вполне рутинной работой для хорошего диплома, тем более в Японии.
Дело в том, что постараться действительно нужно было. До него было несколько попыток определить стереохимию аллильного замещения, но результаты получились, мягко говоря, неоднозначные. Хаяси и сотрудники постарались найти комплекс, с помощью которого стереохимия оказалась определена. И то не со стопроцентной определенностью. Они взяли не очень чистый исходный ацетат со всего 58%-ной оптической чистотой (79% S и 21% R). И получили продукт с 38%-ной оптической чистотой (69% S и 31% R) – то есть ещё 10% по дороге рацемизовались. В наше время, кстати, чёрта с два приняли бы такую статью в джакс – заставили бы дочистить исходное до более высокой энантиомерной чистоты и разобраться с рацемизацией.
Откуда рацемизация, и почему проблемы в аллильном замещении? В первую очередь, потому что η3-аллильные комплексы самопроизвольно и обратимо превращаются в η1-аллильные комплексы, из-за чего палладий, во-первых, блуждает между первым и третьим атомами аллила, а во-вторых, палладий может переползать с одной стороны плоскости аллила на другую, ведь в η1-аллильных комплексах верха и низа нет, они эффективно плоские. Это и есть рацемизация в чистом виде, потому что в этом процессе верх и низ меняются местами и становятся неразличимы. Для простоты и общности опустим другие лиганды на палладии.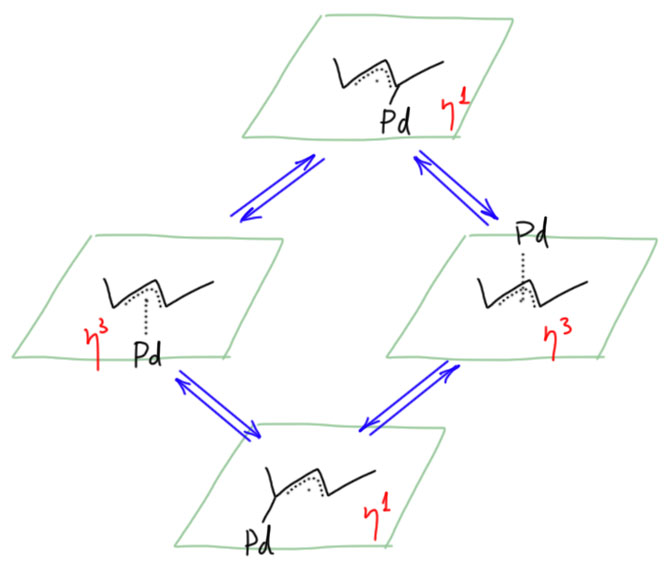
Скорости и равновесия этих процессов зависят от всего на свете, но прежде всего, от других лигандов на палладии. Хелатный фосфин у Хаяси сильно замедлил этот процесс и позволил доказать стереохимию. Монодентатные и более простые лиганды, наоборот, этому процессу способствуют. В целом, здесь всё очень сложно, и использование стереохимии аллильного замещения требует нестандартных решений и нетривиальных подходов. Но это как раз очень интересно для исследователей.
Вопрос. Метод Эчаваррена.
Испанский химик Антонио Эчаваррен, известный очень большим вкладом в применение переходных металлов в органическом синтезе, начинал свою блестящую научную карьеру в лаборатории Джона Стилле. Ему очень понравилась реакция Цудзи-Троста и он решил как-то ее соединить с оловоорганической химией своего руководителя. Получилось очень интересно (A.M.Echavarren, D.R.Tueting, J.K.Stille J. Am. Chem. Soc. 1988, 110, 4039). Поскольку в те ранние времена многое ещё было не очень хорошо понятно, он считал оловоорганику просто нуклеофильным реагентом, посчитав, что она может просто участвовать в каталитическом цикле аллильного замещения. В принципе, у него всё получилось, но с некоторыми фокусами. Фокусы в химии – всегда хорошо, потому что раскрывают новые возможности реакций и дают ключ к тому, что в них происходит на самом деле. Во-первых, хорошо реагировать у него согласился только комплекс палладия без фосфиновых лигандов, в то время как в обычной реакции Цудзи-Троста фосфины всегда желательны, без них каталитический цикл неустойчив. Во вторых, с уже знакомым нам моноэпоксидом циклогексадиена у него получилось два продукта, и оба не вполне такие, которые можно было бы ожидать в нормальной реакции Цудзи-Троста.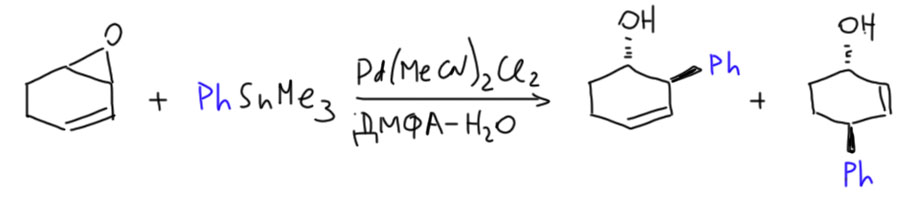
В дальнейшем Эчаваррен сделал из этой реакции неплохой метод получения циклических продуктов (A.M.Echavarren et al. J. Org. Chem. 2001, 66, 589), например: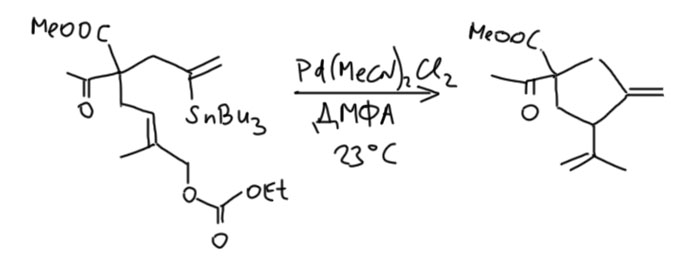
Попробуйте разобраться, как работает метод Эчаваррена. Является ли этот процесс каскадом, и если да, то каким.
(до 20 баллов за наиболее полный и убедительный анализ)
Энантиоселективное аллильное замещение
Наиболее важное применение каталитического аллильного замещения – энантиоселективная реакция, описанная Б. Тростом. Именно асимметрическая версия аллильного замещения вывела эту реакцию в число наиболее популярных методов органического синтеза. Задача эта очень непростая, потому что аллильная система в комплексе находится в системе равновесий моногапто-тригапто-комплексов, в которых аллил переворачивается не только в вертикальной, но и в горизонтальной плоскости. Такие процессы перемешивают все возможные стереохимические конфигурации интермедиатов и продуктов, и никак не способствуют стереоселективной реакции. Катализатор энантиоселективного аллильного замещения поэтому должен не только эффективно различать направления атаки нуклеофила, но и надежно удерживать аллильный лиганд и препятствовать его эпимеризации. По этой причине для этой реакции создают специальные лиганды, не только создающие асимметрическое окружение реакционного центра, но и ориентирующие аллильный лиганд в “хиральном кармане”.
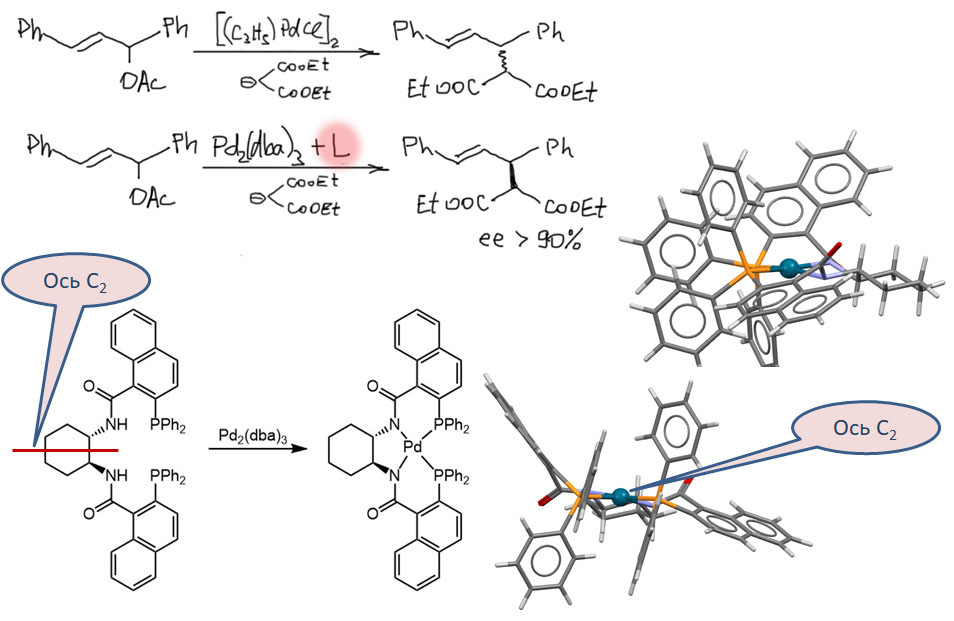
Как очень часто бывает в катализе комплексами переходных металлов, реальная картина действия катализатора оказывается сложнее первоначальной идеи. Вот и в случае энантиоселективного катализа с участием лигандов Троста, было выяснено, что палладий связывается совсем не так, как это рисовала первоначальная красивая модель, предполагавшая связывание двумя фосфиновыми центрами, возможно с поддержкой через амидные лиганды, то есть предполагалось координационное окружение или PdP2 или PdP2N2, как показано на слайде. У такой гипотезы есть один существенный изъян – если комплекс образуется через два фосфора, то это макроциклический хелат – 13 атомов в цикле – а это не очень выгодно и маловероятно. Если предположить, что комплекс образуется с PdP2N2 координацией, то эта проблема отпадает – хелатные циклы получаются оптимальными (их там три штуки, шести-пяти-шести-членных), и такой комплекс действительно с удовольствием образуется, как и показано на реальной структуре на слайде. Но вот беда – координационных мест свободных у него нет для образования аллильного комплекса. В работе Аматоре и сотр. много позже (C.Amatore, J.Organomet.Chem., 2007, 692, 1457) действительно было обнаружено, что в этой системе образуется комплекс совсем другой структуры – простой хелат, в котором палладий связан с фосфором и кислородом, и такой комплекс может включать и аллильный лиганд. Вот он, на картинке. Учтите только, что палладий в этом комплексе имеет степень окисления +2, и другого и быть не может, потому что никаких окислительно–восстановительных реакций при образовании этого комплекса не происходило, комплекс образовался, как положено, просто за счет равновесий лигандного обмена. Следовательно, палладий в нем имеет формальный заряд +1.
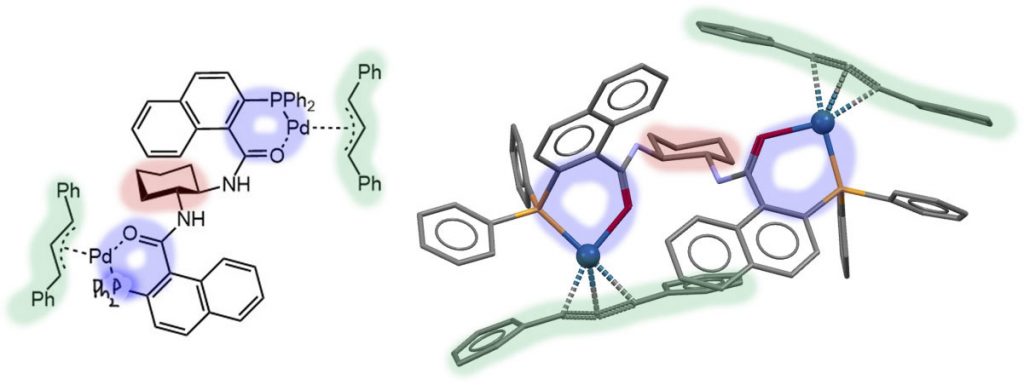
Красиво? Невероятно. Но вот как такой комплекс может участвовать в переносе хиральности – аллильный комплекс в нем полностью выведен из асимметрического окружения, которое обеспечивает транс-диаминоциклогексановый остов лиганда. Посмотрите – где аллил, а где хиральность. Но ведь работает эта каталитическая система.
Получается довольно головоломная загадка. И очередная иллюстрация, как красивая идея двигает экспериментальные исследования, и эти исследования дают реальный результат в виде селективных и эффективных протоколов. И все радуются, потому что основная цель химии – не писать красивые теории, а получать новые интересные соединения. И когда начинают разбираться, как же работает уже найденная система, очень часто оказывается, что первоначальная красивая теория очень далека от реальности.
Вопрос. Каскад Хек/Цудзи-Трост.
Попробуйте объяснить, как работает каскад Мидзороки-Хек/Цудзи-Трост. Объясните, почему это именно настоящий каскад, а не тандем, как иногда пишут в статьях про эту реакцию. Напоминаю, что тандем – это два или больше соединённых циклов, со своим катализатором в каждом (катализатор следующего цикла может образоваться из катализатора предыдущего, но это другой катализатор), а каскад – один цикл, связанный одним катализатором. Эта терминология не сразу устоялась, поэтому в старых работах эти понятия часто используют беспорядочно.
R. C. Larock, Y. D. Lu, A. C. Bain and C. E. Russell, J. Org. Chem., 1991, 56, 4589–4590
(до 20 баллов)
Пример: Два реагента из одного исходного. Оксазолиновые лиганды.
При образовании аллильного комплекса из карбонатов образуется алкоксид из второй группы карбоната. Цудзи быстро придумал ещё более изощрённый способ использовать эту реакцию (Tetrahedron Lett.,1983, 24(l7), 1793). А что если с другой стороны не просто алкоксид, а енолят. Тогда образовавшийся енолят тут же станет нуклеофилом в реакции с аллильным комплексом. Такие карбонаты легко получаются обычной енолятной химией с помощью аллилхлороформата (хлороформаты, так странно вполне устойчивые, но бешено реакционноспособные называются полуэфиры полухлорангидриды угольной кислоты потому что неустойчивый полухлорангидрид угольной кислоты ClCOOH издревле называют хлормуравьиной кислотой). Получим, например, такое производное из циклогексанона: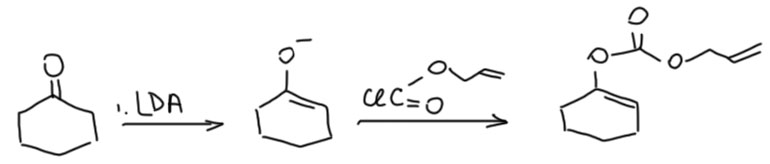
Обрабатываем этот карбонат палладиевым пред-катализатором. Карбонат тут же разваливается на енолят и аллильный комплекс, которые друг с другом и реагируют: 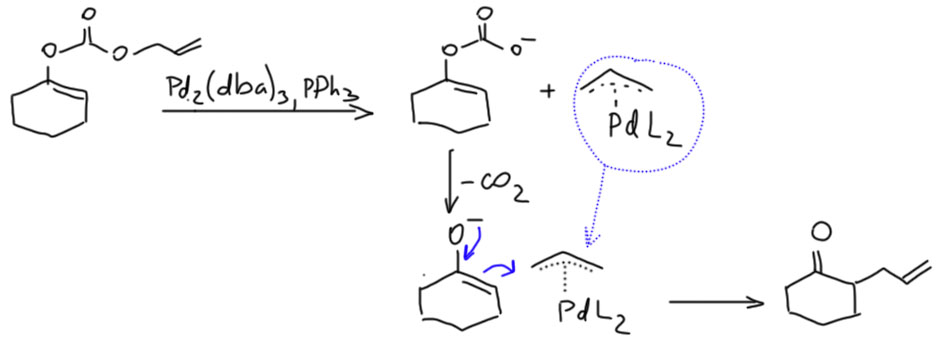
И хотя конкретно этот продукт сильно не впечатляет – мы на 3 курсе его получим без проблем, но это не единичный пример, а очень мощный синтетический метод.
Развитие этого метода, естественно, пошло по пути энантиоселективности. Биэнна и Штольц использовали метод для энантиоселективного аллилирования, но не со стороны аллильной системы, как это делают обычно, а со стороны енолята, то есть взяли прохиральный нуклеофил, енолят (D.C.Behenna, B.M.Stoltz J.Am.Chem.Soc. 2004, 126, 15044). Как всегда в таких исследованиях, экспериментально подбирают хиральный лиганд, чтобы получить побольше и выход и оптическую чистоту. Лиганд Троста, с которым мы уже знакомы сработал неплохо, но ee было меньше 70% (это вполне приличный результат, но хочется большего). BINAP провалился полностью – с ee в 2% – убеждаемся, что знаменитый лиганд в конкретной реакции вовсе не гарантия успеха. Отличный результат получился с хелатным лигандом изоксазолинового ряда. Мы впервые сталкиваемся с этими лигандами, а это одно из самых эффективных семейств лигандов в энантиоселективном катализе. Хиральные оксазолины очень легко получаются из разнообразных нитрилов и хиральных аминоспиртов, которые элементарно получаются из оптически чистых природных и синтетических аминокислот. Это целое огромное семейство лигандов, – и нитрилов разных много и аминокислот, чрезвычайно популярное в асимметричеком синтезе. В группу R’ можно запихать что угодно, например, другие координационные центры для создания хелатного лиганда. Сразу скажу, что когда оксазолиновый цикл один, его асимметрия определяется только одним стереогенным центром, и это не очень сильная асимметрия, чтобы можно было ожидать высокой оптической чистоты в разных реакциях. Гораздо более эффективны лиганды с двумя такими кольцами, создающими при хелатировании C2-симметричное окружение атома металла – их обычно называют BOX-лигандами (bis-oxazolines). 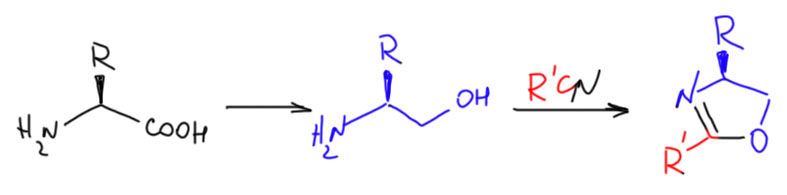
Но в этом случае вполне сработал моно-оксазолиновый лиганд с фосфиновым заместителем. Высокую оптическую чистоту обеспечивает объемистый заместитель на стереогенном центре.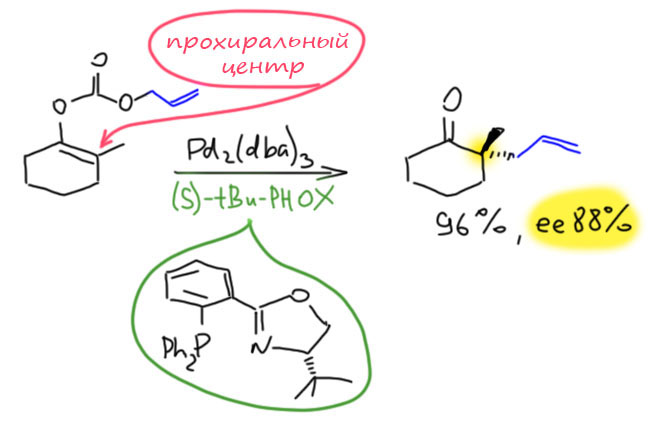
Генерация активных монолигандных катализаторов
Аллильные комплексы оказались очень удобны для генерации монолигандных комплексов, так как в пред-катализаторе как раз умещается один аллильный лиганд и один полезный лиганд (Marion, M.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4104). Если в реакционной смеси есть какой-нибудь нуклеофил, как, например, в реакциях Судзуки-Мияуры или Бачуолда-Хартвига, то такой пред-катализатор легко за счет аллильного замещения избавляется от аллильного лиганда, и остается с одним полезным лигандом. В качестве временного лиганда в таких пред-катализаторах обычно используется не самый простой аллил, а его гомолог – метилаллил или кротил, по причине более высокой скорости активации, хотя, как обычно, опыт применения в разных ситуациях показал, что и аллил иногда бывает хорош. Это вообще обычная история в химии – сначала вы что-то долго исследуете и находите наилучший вариант, долго объясняете, почему именно этот вариант совершенно незаменим, используя самые изощрённые доводы, квантово-химические расчёты высочайшего уровня, и всё такое. В общем, всем ясно, что вы открыли новый закон Природы. А через некоторое время выясняется, что бывает и не так, и все ваши высокоумные объяснения повисают в воздухе, как улыбка Чеширского кота.
Это вообще обычная история в химии – сначала вы что-то долго исследуете и находите наилучший вариант, долго объясняете, почему именно этот вариант совершенно незаменим, используя самые изощрённые доводы, квантово-химические расчёты высочайшего уровня, и всё такое. В общем, всем ясно, что вы открыли новый закон Природы. А через некоторое время выясняется, что бывает и не так, и все ваши высокоумные объяснения повисают в воздухе, как улыбка Чеширского кота.
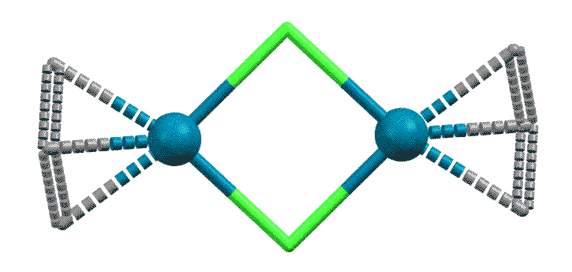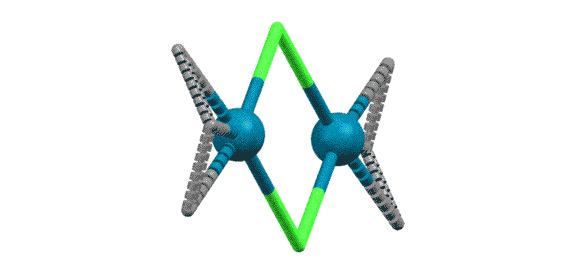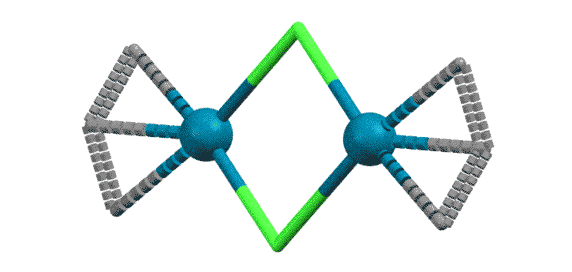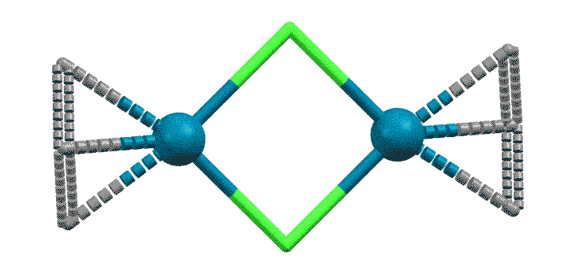
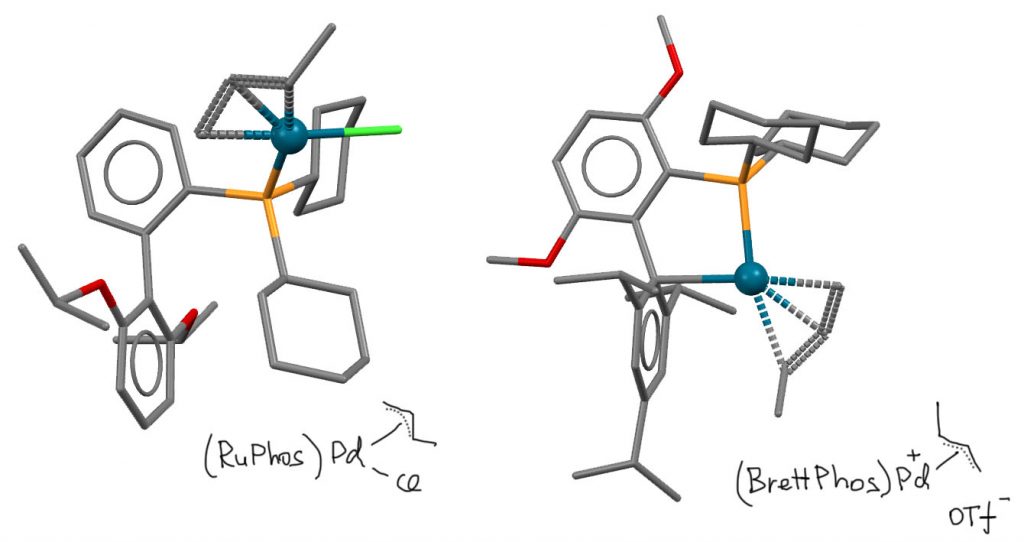
Два таких предкатализатора показаны ниже (T.Colacot et al, J. Org. Chem. 2011, 76, 7918–7932). Структура зависит еще от одного лиганда – или галогенида, или трифлата в зависимости от того, какое аллильное производное использовано. Если это галогенид, структура пред-катализатора вполне очевидно – у палладия три лиганда, фосфин, аллил и галогенид. Если это трифлат, то он не сидит в координационной сфере, а служит просто противоионом, комплекс в этом случае имеет формальный положительный заряд, а палладий ищет дополнительный лиганд, прицепляясь ко второму кольцу дифенильного остова фосфина. Специфика лигандов с диизопропилфенильной группой типа X-phos и BrettPhos состоит в том, что палладий не может уцепиться дигапто-способом за двойную связь кольца, и цепляется более слабо за один углерод.
Пример использования предкатализаторов аллильного типа
Универсальных методов не бывает. И если алифатические и ароматические первичные амины довольно легко реагируют в реакции C-N кросс-сочетания, циклопропиламин страдает от больших проблем. Долгое время для арилирования циклопропиламина хороших протоколов не было. Сочетание лигандов третьего поколения с пред-катализатором аллильного типа дало наконец хороший протокол арилирования циклопропиламинов ( T. J. Colacot et al. Org. Lett. 2016, 18 (6) , 1442-1445). Для бромпроизводных используется очень рогатый лиганд дифенильного ряда tBuBrettPhos.
Если арилбромид тоже рогами упирается, чуть-чуть ослабляют рогатость лиганда, иначе они забодают друг друга и ничего не получится. Берут простой BrettPhos:
И с хлорпроизводными работает, только немного пожёстче условия нужны