
Back donation
Когда мы сосредоточиваем внимание на 18-электронном правиле и его следствиями, мы представляем себе любое координационное соединение очень просто – связь металла с лигандом всегда координационная: металл выступает как кислота Льюиса, а лиганд как основание Льюиса. Смещение электронной плотности в таком взаимодействии происходит от лиганда к металлу. Это всегда так, даже в комплексах низковалентных поздних переходных металлов. Как это можно представить, мы уже разобрали.
Такое представление дает очень удобный рецепт быстрой оценки того, правильно ли мы поняли состав комплекса, является ли он координационно насыщенным, и способен ли принять еще лиганды. Но если мы захотим понять, как тот или иной комплекс реально устроен, этого представления может оказаться мало.
В чистом виде картинка связывания, описываемая 18-электронным правилом – один лиганд – одна орбиталь, – может быть только у гидридных комплексов, так как у атома водорода действительно больше ничего нет кроме одной s-орбитали. Но для любых других лигандов совершенно понятно, что это только малая часть. Если вы когда-нибудь видели результаты квантово-химического расчета любого негидридного комплекса и любым методом, кроме старого и очень доброго расширенного метода Хюккеля, то не могли не заметить, что только число занятых МО во много раз больше, чем (9+число лигандов) – то мы рисовали, обсуждая 18-электронное правило. Это не может быть по другому, потому что даже если мы сможем выделить из набора орбиталей только те, что относятся к валентным оболочкам атомов, и ограничимся только занятыми, то каждый неводородный атом лиганда даст их не менее 2 штук, итого будет очень много. Сильно волноваться по этому поводу не стоит, подавляющее большинство этих МО относятся только к лигандам, с орбиталями металла не взаимодействуют, в связывание с металлом ничего не дают, и их можно без ущерба назвать с точки зрения структуры комплекса несвязывающими. Не стоит думать, что они совсем бесполезны, напротив, для многих свойств комплекса, например, всяких спектров, оптических свойств, электрохимии, и т.п. они могут оказаться даже важнее, чем орбитали, участвующие в связывании. Но мы этим не занимаемся, а с точки зрения устойчивости комплекса и его реакционной способности это не так важно.
Тем не менее, и связи металла с лигандами часто не так просты, как это подразумевает 18-электронная модель. Попробуем разобраться, сначала на нескольких типичных примерах.
Комплексы оксида углерода
Оксид углерода не только один из самых важных лигандов, но и просто идеальная модель для изучения взаимодействия металл-лиганд. Это очень маленькая молекула, электронное строение которой известно до мельчайших деталей. В терминах молекулярных орбиталей эта двухатомная молекула располагает очень понятным набором орбиталей, из которого для связывания с переходным металлом мы до сих пор использовали только одну. Какую? Есть смысл немного подробнее разобраться с тем, как это устроено. Сама молекула оксида углерода имеет хорошо известную структуру. Формально это соединение двухвалентного углерода, но в этом случае атом углерода имел бы только 6 валентных электронов, был бы секстетным атомом, а оксид углерода был бы … карбеном. В принципе, это не такое страшное слово, и так могло бы быть, и мы еще увидим множество стабильных карбенов, имеющих прямую и очень интересную связь с этой формой оксида углерода. Но все-таки это невыгодно, и поэтому атом кислорода охотно делится одной парой. Соответствующая граничная структура имеет sp-гибридные атомы углерода и кислорода, тройную связь, точно такую же как в молекулах ацетилена или цианидов (цианид-ион полностью изоэлектронен и изоструктурен оксиду углерода, как, впрочем и дианион ацетилена). Реальная молекула оксида углерода практически идеально описывается именно этой граничной структурой, в которой нет секстетных атомов, в которой кислород имеет формальный положительный, а углерод формальный отрицательный заряд (в реальности эти заряды компенсируются поляризацией ковалентных связей в сторону более электроотрицательного атома кислорода, и суммарные заряды на атомах практически близки к нулю).
Теперь посмотрим на молекулярные орбитали. Сначала занятые. Это очень просто, тем и хороши такие простые молекулы. Смотрим на левую граничную структуру. Неподеленные пары на кислороде и углероде – две МО. Сигма-связь – еще одна МО. Две одинаковые π-связи – еще две МО, причем одинаковые, одинаковой энергии, вырожденные. Итого пять МО, три имеют σ-тип (расположены вдоль оси связи), и две π-тип. Поскольку три σ-МО образованы sp-гибридными орбиталями атомов, они почти одинаковы по форме, но в одной максимум плотности находится на внешней доле у атома кислорода (это соответстует неподеленной паре на кислороде), у второй между атомами (это в основном σ-связь, а в третьей – на внешней доле у атома углерода, соответствуя неподеленной паре на углероде. Именно эта последняя является самой высокой занятой МО (ВЗМО). Понятно почему – это фактически несвязывающая орбиталь, и расположена она там, где обычно располагаются несвязывающие орбитали. Так, а почему тогда там же нет той орбитали, которая соотвествует паре на кислороде – это же тоже несвязывающая? Прежде всего потому что кислород обладает очень высокой электроотрицательностью, а это соответствует относительно низкой энергии всех занятых орбиталей, локализованных на таком атоме, или хотя бы имеющих большую плотность на таком атоме. Поэтому эти орбитали погружаются глубоко, опускаются ниже связывающих π-МО. Это очень важная особенность. С виду молекула CO симметрична, и пары обозначены с обеих сторон, но та, что у углерода намного более доступна для связывания с металлами. Это и есть то, что по-другому называют теорией ЖМКО, и то, что описывают как мягкое, соответствует именно максимально выгодным орбитальным взаимодействиям – орбитальному контролю. Следовательно, оксид углерода – мягкий лиганд, а точнее, мягкое основание Льюиса, и центр мягкого связывания находится на атоме углерода. И даже еще точнее, поскольку связывание осуществляется за счет орбитали σ-типа и у металла тоже будет выбрана d-орбиталь с подходящей симметрией (dσ), то оксид углерода можно назвать σ-основанием Льюиса. А металл в этом взаимодействии работает как σ-кислота Льюиса.
Сразу под ВЗМО находится пара одинаковых, но взаимно ортогональных π-связывающих орбиталей. Они выглядят точно так же, как такие же орбитали тройной связи в ацетилене с одной разницей – веса орбиталей на атомах сильно различаются, на атоме кислорода больше плотности. Это вполне очевидный эффект – связи поляризованы в сторону кислорода, и это и есть молекулярно-орбитальное описание этой поляризации. И вообще, нас эти орбитали пока не очень волнуют, металл ведь мы уже привязали и сейчас просто балуемся с оставшимися орбитальками. Вот еще на самые низколежащие незанятые МО (НСМО) посмотрим. Соверешенно очевидно, что это орбитали, парные к связывающим π-МО, а именно π-антисвязывающие. А почему нет парной к σ-ВЗМО? А потому что она несвязывающая, у несвязывающих МО нет явной пары. И эти антисвязывающие орбитали, π*-НСМО естественно очень похожи на свои парные связывающие, построены-то они из одного материала, только доли вывернуты, чтобы между ними была узловая плоскость, и – там где на связывающей больше у кислорода, у антисвязывающей – у углерода. И вот это-то нам сейчас и пригодится…
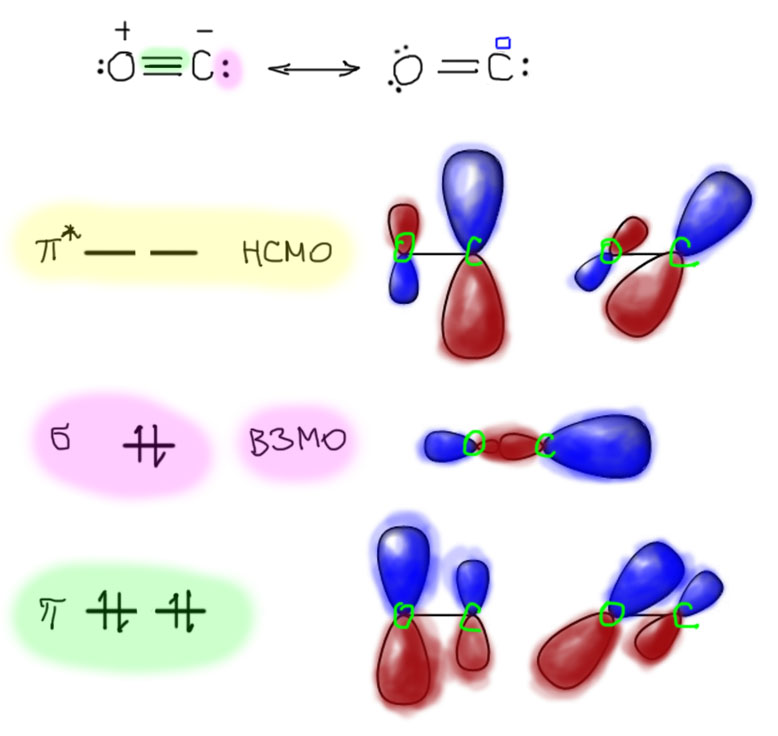
Металл мы уже привязали σ-связью. И теперь представим, а что если у него остались еще d-орбитали с электронами поверх тех, что уже использованы для связывания. То есть несвязывающие, ровно те самые, которые мы уже видели, когда считали электроны и пытались понять, как объяснить 18-электронное правило. В связывании обычной координационной связью задействованы dσ-орбитали, а dπ-АО пока остаются без дела. Нарисовав такую dπ-орбиталь, мы замечаем, что на атоме углерода, с которым этот металл уже связан, есть p-орбиталь, входящая в состав антисвязывающей π*-НСМО. Почему антисвязывающей, там же есть еще связывающая π-МО? Есть, но во-первых, там тоже есть два электрона, а две занятые орбитали не взаимодействуют, а во-вторых, на этой связывающей, как мы уже выяснили, доля p-орбитали углерода мала, а вот на антисвязывающей наоборот велика. И взаимодействие заполненной dπ-АО металла и пустой орбитали связи выгодно, так как их симметрия друг другу подходит (синенькое на синенькое, красненькое на красненькое). Конечно, нужно еще знать их относительное расположение, близки ли они по энергии? Насколько близки – это зависит от металла и электронной структуры конкретных лигандов, но точно не очень далеки, потому что и одна и другая находятся сверху пачки связывающих орбиталей, прямо сразу после тех занятых орбиталей, которые уже использованы.
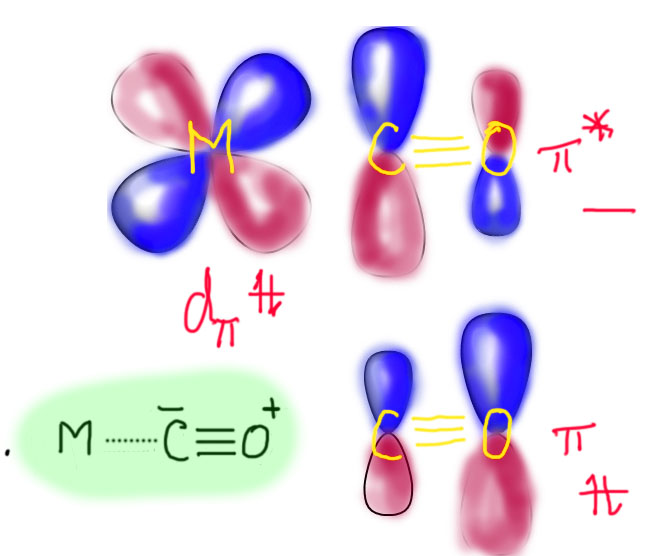
Back-donation: взаимодействие с переменным порядком связи
Извиняюсь за чрезвычайно корявый и мутный заголовок, но ничего лучше придумать не мог.
Проблема в том, что для связи, которую образует металл за счет занятой dπ-АО и лиганд за счет вакантной разрыхляющей π*-НСМО, нет нормального обозначения в русском языке. По-английски это называется back-donation, и как это будет по-русски? А также по-французски, немецки, испански, и т.п. Никак. Английский язык потрясающе хорошо приспособлен для научной речи как минимум потому что слова можно склеивать без служебных грамматических прокладок, а всем остальным проще просто взять английский термин и использовать как есть. В других европейских языках с латинской графикой никто и не заметит, что это что-то чужое, но в языках со своей графикой такие термины зрительно вываливаются из потока букв. Но ничего, привыкнем, и мне не очень хочется искать русский аналог, уж больно они все корявые. Итак, пусть будет back-donation.
Вторая проблема гораздо более серьезна. Back-donation – это химическая связь, или какой-то эффект, типа индуктивного, приводящий к небольшому смещению электронной плотности в рамках уже существующей связи? А это вообще важно?
Важно. В химии понятие структуры – одно из самых фундаментальных. И в любой структуре мы умеем выделить химические связи, и распознать всякие вторичные эффекты, приводящие к значимому перераспределению электронной плотности. Распознаются связи достаточно надежно, во-первых, по межатомным расстояниям: каждому типу связи соответствуют свои характерные величины расстояний, очень хорошо сохраняющиеся с довольно маленьким разбросом значений в разных соединениях. Именно так распознают связи, например, программы, которые обслуживают рентгеноструктурный анализ – этот метод не знает ничего про электронную структуру и химические связи в квантово-химическом смысле, а просто определяет положения атомов в пространстве, а значит и расстояния между ними, но на картинках уверенно рисует связи, и очень редко ошибается. Во-вторых, по данным квантово-химических расчетов электронной структуры находится распределение электронной плотности в молекуле, и через различные схемы деления объема молекулы на отдельные атомы (это называется partitioning) определяется доля плотности в совместном владении пар атомов. Эти схемы обычно дают нечто, чрезвычайно похожее на то, как мы рисуем структурные формулы, обозначая связи черточками, и утверждая, что каждая черточка обслуживается парой электронов. В случае делокализации, многоцентровых связей и тому подобных фокусов, доля плотности между вовлеченными в эти дела атомами будет заметно меньше пары (или двух, или трех пар, если изображаемые связи кратные), и так далее, главный вывод из всего этого таков, что старая добрая структура, изображаемая черточками связей и точечками неподеленных пар, и даже картинками мезомерной делокализации очень неплохо отражается в данных квантово-химических расчетов. И в случае координационных соединений эта картина будет очень похожа на ту структуру, которую мы рисуем для подсчета электронов в 18-электронном правиле. Для координационных соединений с лигандами с гаптностью больше 1 придется это учесть, принимая, что плотность, отвечающая за связи с таким лигандом не двух, а многоцентровая.
Back-donation становится настоящей связью. Или нет?
Как можно понять, работает ли back-donation и в какой степени, и может ли этот эффект достигнуть такой мощи, что не заметить это будет нельзя, и придется призадуматься, а не настоящая ли это химическая связь?
Оксид углерода (карбонил) – очень удобный лиганд для такой демонстрации. Известны его комплексы почти со всеми переходными металлами в самых разных окислительных состояниях. Вот, посмотрим, например, на три практически совершенно одинаковых комплекса металлов 8-10 групп с конфигурацией d10 и тетраэдрической координацией. Единственная разница – заряд, необходимый для поддержания этой электронной конфигурации. Вот они, по данным РСА (противоионы и всякий мусор, застревающий в кристаллических решетках, опущен). Кроме цвета центрального шарика, кажется, совсем одинаковы. Размеры шариков довольно заметно уменьшаются к никелю, и это не случайно – хоть эти шарики и не имеют никакого отношения к размерам атомов, что бы под этим ни понимать, и рисуются чисто для красивой картинки, эти размеры специально подбирают пропорциональными известным атомным размерам, и это видимое сокращение вполне отражает сжатие атомов в периодах из-за увеличения заряда ядра.
Но посмотрим на длины связей, и увидим интересную тенденцию – связь C-O в карбониле заметно уменьшается от железа к никелю, а связь металл-углерод, напротив, увеличивается. Второе даже более странно – размер атома-то уменьшается. Можно предположить, что по каким-то причинам в этом ряду связь с лигандом ослабевает, а связь атомов в CO, наоборот, крепнет.
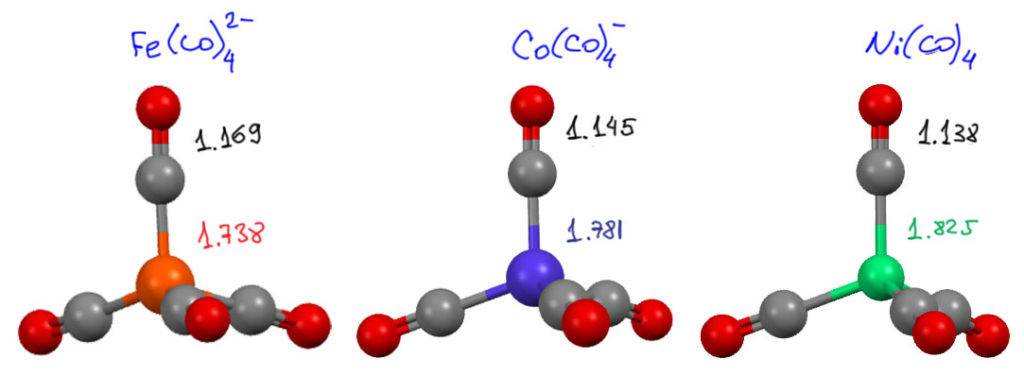
Вопрос первый: back-donation есть во всех этих комплексах, или нет? Почти наверняка во всех, как минимум потому что в конфигурации d10 нет недостатка в электронах для этого эффекта. Кроме того, даже в карбониле никеля длина связи в карбониле заметно больше, чем в молекуле CO (1.128 Å), а если эта молекула связана с металлом только координационной σ-связью, вообще не имеющей прямого отношения к орбиталям, обслуживающим связь углерода и кислорода, то связь C-O либо совсем не должна изменяться, либо даже может немного ещё уменьшаться из-за частичного переноса электронной плотности с σ-орбиталей на металл.
Вопрос второй: а к чему приводит back-donation в карбонильных комплексах с таким типом координации? Очень просто – если представить себе, что из пары электронов на dπ-орбитали металла один целиком перешел на карбонил, на разрыхляющую орбиталь, то это можно просто изобразить. Электрон на разрыхляющей орбитали, соотвествующей одной из π-связей карбонила, ослабит эту связь, сделав ее 3-электронной и следовательно разорвав ее, причем пара электронов отойдет кислороду, а один – углероду. Ставим формальные заряды, соотвествующие перемещению электрона, и обращаем внимание на то, что на металле и углероде образовалась спаривающаяся, связевая пара, и это и есть настоящая связь, соотвествующая back-donation. Эффект очевиден – связь CO ослабнет, станет более длинной, а связь металл-углерод наоборот станет более прочной, сократится.

Вот ровно это мы и видим в ряду карбонилов. Тетракарбонил никеля в этом смысле – точка отсчета, в нем есть некоторая степень back-donation, и это приводит к небольшому увеличению длины связи CO (и еще сказывается на частоте колебаний этой связи в ИК-спектре, но пока отложим этот непосредственно связанный с длиной связи эффект). В тетракарбонилкобальтат-ионе степень участия back-donation повышается. Почему? – и потому что на металле формально отрицательный заряд, да и просто потому, что d-орбитали несколько выше, чем у никеля (еще раз вспомним, что уровни энергии всех АО металла понижаются от начала к концу ряда). Мы даже можем сказать, что эффект back-donation перераспределяет электронную плотность с металла на лиганд, поэтому, видимо, минус на металле там формальный, липовый, а реально минус расползся по атомам лигандов. Ну и дианион тетракарбонилата железа – совсем уже яркий пример – еще сильнее back-donation, еще слабее связь CO (по ИК это уже почти просто настоящая двойная связь, а не тройная, хотя по длине еще далековато). И, видимо, и тут тоже минусы с металла расползлись по атомам неметаллов в лигандах. И, естественно, добавление все большего вклада back-donation связывания к уже существующим координационным связям делает суммарно связь металл-лиганд все прочнее и короче именно от никеля к железу, а не наоборот, как можно было бы ожидать из размеров атомов.
Итак, этот поучительный кейс показывает, что back-donation – это все-таки не полноценная химическая связь, а именно такой переменный π-довесок к основной координационной σ-связи.
Впрочем, вот еще один кейс, теперь из химии ранних переходных металлов.
Back-donation в химии ранних переходных металлов. Всё-таки становится?
Как мы уже выяснили, ранние переходные металлы щедро раздают всем свои электроны, и почти всегда остаются с пустыми полками валентных оболочек и конфигурацией d0. На back-donation там нет ничего, и говорить не о чем. Ранний переходный металл в обычной для себя максимальной степени окисления – это просто кислота Льюиса, приемный пункт электронных пар от лигандов.
В 3 группе это всегда так. Но начиная с четвертой появляются кое-какие интересные варианты. Возьмем, например, титан, важнейший элемент в томй химии, которую мы взялись изучать. Координационная химия титана и реакции с органическими молекулами разнообразны и изумительны, иногда необычны настолько, что вызывают ощущение какого-то невозможного бреда.
Титану свойственны не только нормальная степень окисления Ti(4+), но и несколько промежуточных, особенно часто попадается Ti(2+). Добавим к этому то, что мы уже обсуждали – у самых ранних переходных металлов, видимо, почти совсем недоступны p-оболочки, и они играют в координационную химию только с помощью sd-оболочки с 12 местами, а дополнительные лиганды принимают за счет повышенной ионности связей с помощью способа связи, напоминающего гипервалентность неметаллов 15-18 групп (короткопериодных 5А-8А подгрупп). Мы уже видели, что у титана есть 14 и 16-электронные комплексы. А 18-электронные? Да, сколько угодно. Это не основное состояние этого металла и вообще металлов 4 группы, но оно хорошо представлено, и очень важными комплексами с карбонильными лигандами. Просто карбонил титана, продолжающий ряд гомолептических карбонилов, и заканчивающийся на 17-электронном карбониле ванадия V(CO)6, а у него должна была бы быть формула Ti(CO)7, так до сих пор и не был получен. Но известны и вполне однозначно охарактеризованы и бис-дифосфиновый комплекс, в котором из исходного карбонила четыре замещены на донорные фосфины, и это 18-электронный комплекс Ti(0). И есть даже анионный комплекс с формальной степенью окисления Ti(2-). И, наконец, очень хорошо изученный комплекс – дикарбонильное производное титаноцена. Эти три комплекса образуют очень интересный ряд, в котором последовательно растет формальная степень окисления металла и координационное число, с одновременным уменьшением числа d-электронов. При этом длина связи CO явно завышена относительно длины тройной связи, и у одного из комплексов даже очень сильно, но никаких четких и понятных тенденций не просматривается.
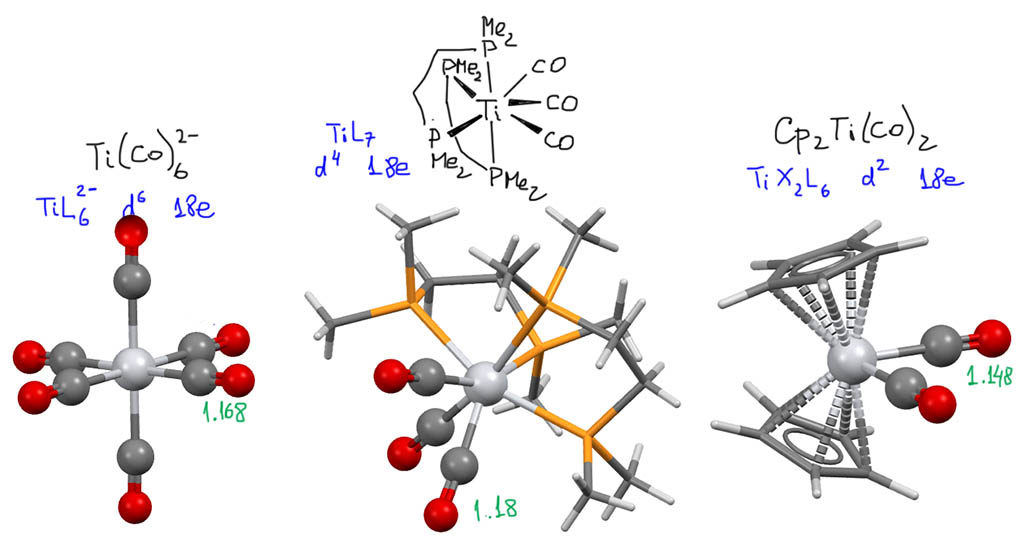 Глядя на этот ряд, начинаешь невольно сомневаться, так ли хорошо описывают комплексы титана, а возможно и не только их (как минимум, в 4 группе все металлы дают близкие по структуре соединения), все эти степени окисления и числа d-электронов, и все остальные формальные параметры. В этом, на самом деле, нет ничего страшного, если не забывать, что эти величины и характеристики именно таковы и есть – формальные. А реальная структура, скорее всего, немного другая. Возьмем, например, последний комплекс. Образование этого комплекса можно представить, как реакцию титаноцена с двумя молекулами CO. Титаноцен – точно комплекс Ti(2+), а это – очень сильный восстановитель. Ранний переходный металл действительно предпочитает максимальную степень окисления 4+, поэтому все состояния с меньшими степенями окисления мечтают сбагрить свои электроны первому попавшемуся. Попалась молекула CO – тот еще окислитель, но для Ti(2+) вполне. И если мы посмотрим на реакцию образования бис-карбонильного комплекса именно как на редокс-реакцию.
Глядя на этот ряд, начинаешь невольно сомневаться, так ли хорошо описывают комплексы титана, а возможно и не только их (как минимум, в 4 группе все металлы дают близкие по структуре соединения), все эти степени окисления и числа d-электронов, и все остальные формальные параметры. В этом, на самом деле, нет ничего страшного, если не забывать, что эти величины и характеристики именно таковы и есть – формальные. А реальная структура, скорее всего, немного другая. Возьмем, например, последний комплекс. Образование этого комплекса можно представить, как реакцию титаноцена с двумя молекулами CO. Титаноцен – точно комплекс Ti(2+), а это – очень сильный восстановитель. Ранний переходный металл действительно предпочитает максимальную степень окисления 4+, поэтому все состояния с меньшими степенями окисления мечтают сбагрить свои электроны первому попавшемуся. Попалась молекула CO – тот еще окислитель, но для Ti(2+) вполне. И если мы посмотрим на реакцию образования бис-карбонильного комплекса именно как на редокс-реакцию.
В этом случае можно считать, что при образовании комплекса низковалентный титан восстанавливает лиганд, сам переходя в обычную для переходного металла степень окисления Ti(+4), но при этом с зарядом на металле +2, а на карбонилах в сумме -2. Более того, в этом случае возможна ситуация, когда карбонилы привязаны к металлу не координационной связью – ее просто нет, а только связью back-donation. C точки зрения счета электронов получается немного парадоксальная ситуация – лиганды есть, а электроны на это не выделяются, и комплекс, c точки зрения счета электронов, приобретает вид Cp2Ti(+4) c конфигурацией металла d0, структурным типом MX2L4 и счетом электронов 0 + 6×2 = 12!!!! Мы ловко сделали из 18-электронного комплекса 12-электронный и глазом не моргнули. Жульничество чистой воды! Полиция!! Титан ограбили!!!
Пусть полиция займется настоящими жуликами. Реальная структура этого комплекса показывает, что такое описание очень близко к истине. Расчеты и некоторые экспериментально измеримые характеристики структуры говорят о том, что степень окисления металла действительно, видимо, +4; что на металле действительно сидит большой положительный заряд, уравновешиваемый отрицательным зарядом на лигандах; что способ связи лигандов и металла действительно сильно напоминает картину гипервалентности, с которой мы уже разбирались, когда выясняли, как самые ранние переходные металлы справляются с проклятием недоступности p-оболочек – иллюзия возможности связывания большего числа лигандов, чем позволяет 12-электронная валентная sd-оболочка, достигается высокой степенью ионности связей.
Из всей этой истории есть два вывода, плохой и хороший. Хороший состоит в том, что у ранних переходных металлов действительно своя собственная химия, и мы еще не раз увидим, как это отражается на их реакционной способности. Плохой – в том, что простая схема счета электронов для ранних переходных металлов работает плохо, а точнее, совсем никак не работает, и что back-donation способен становится настоящей химической связью, даже иногда вовсе заменяя обычную координационную, а иногда дополняя ее, но у нас нет простых рецептов работы с этими структурными фокусами, а структурные формулы вообще перестают отражать реальность. Придется каждый раз разбираться в том, как действительно устроена та или иная структура. В качестве задания можете попробовать проанализировать два оставшихся комплекса титана и выяснить, сколько там на самом деле электронов, и добавить ещё по 4 плюсика.
Внимание, провокация! В тексте этой главки есть довольно глупая ошибка. Кто найдёт и объяснит, в чём она заключается, получит до 20 плюсиков приз сгорел, а ошибка будет исправлена.
Ошибка заключается в том, что back-donation не отменяет координационную связь, а является дополнительным электронным эффектом, что мы собственно везде и утверждаем, и используем. Поэтому карбонилы вовсе не висят только на back-donation, то есть не становятся Z-лигандами (см. далее), они остаются L-лигандами, а если угодно в данном случае LZ-лигандами. Счёт электронов это не изменяет, полицию звать не надо. Счёт электронов не изменяется, потому что электроны для back-donation уже учтены в счёте, только раньше они были формально несвязывающими, а теперь стали связывающими, но остались в счете валентных электронов металла, потому что это и есть валентные электроны металла. И если до того момента, как мы увидели этот эффект, у нас получалось 18 электронов, то столько же их и останется. Текст специально переписывать не буду, потому что на ошибках учиться быстрее и лучше, и зачем терять такую оказию.
Z-лиганды
Мы видим, что лиганд может висеть только на back-donation. Это может быть самый обычный лиганд, который в других комплексах работает как нормальный L или X-лиганд. В этих случаях, мы не меняем классификацию лиганда, а просто внимательно анализируем ситуацию и пытаемся понять, как обеспечена связь металла и лиганда в конкретном комплексе. Но, хоть и очень редко, но может быть ситуация, когда лиганд в принципе не имеет возможности связываться ни как L, ни как X, потому что не имеет валентных возможностей – нет у него ни свободной электронной пары, ни даже завалящего валентного электрона.
А что это вообще? Разве такое бывает? Что это за жалкая побирушка, ничего у него нет, ещё пусть скажет, что не ел шесть дней. Да нет, ничего особенного, вполне распространенная ситуация – это же просто описание кислоты Льюиса. Может ли кислота Льюиса быть лигандом? Как-то, на первый взгляд, кажется, что это нонсенс – ведь сам металл в комплексе это и есть по определению кислота Льюиса. Зачем одной кислоте Льюиса другая кислота Льюиса? Мы же так любим повторять мантру “электрофил с электрофилом не реагирует”, вопрос закрыт. Но тут мы вспоминаем про back-donation, означающее, что металл может быть основанием Льюиса и нуклеофилом. Вполне понятно, что это не совсем обычная вещь, и комплексов с таким сочетанием металл-лиганд не может быть много. Но они есть, и как только ими заинтересовались, их число стало множиться, исследователи стали целенаправленно охотиться за такими комплексами, исследовать их свойства и реакции. Сейчас это вполне отдельная и модная тема. Термин Z-лиганды быстро укоренился и сейчас не вызывает сомнений. Пора и нам его освоить. Примечание: этот раздел был написан до 2022 года, и тогда никто не мог даже в диком сне увидеть, что простая буква латинского алфавита может приобрести какой-то странный смысл; и мы не будем плясать под эту дудку и что-то исправлять – настоящая наука сильно выше и долговечнее преходящей дури шарлатанов, и не требует суеты и подстраивания под обстоятельства.
Лиганды такого типа – только кислоты Льюиса, требующие эффективного back-donation от металла – выделили в отдельный класс лигандов и назвали их Z-лигандами. Обычно это производные p-элементов, или чистые кислоты Льюиса, которых много среди соединений бора и алюминия, а также галлия и индия. Или соединения, являющиеся кислотами Льюиса, например, из-за доступных σ*-орбиталей или даже π*-орбиталей, как у силанов или, как ни странно, молекулы SO2, которая вроде бы вполне себе нуклеофил, но с металлами часто взаимодействует именно как Z-лиганд.
Проблема Z-лигандов состоит в том, что они весьма слабы как обычные монодентатные лиганды – всё же такое взаимодействие довольно малоэффективно, в частности, из-за большого размера атомов, вовлеченных в такие связи, а это приводит к большим межатомным расстояниям, что не способствует хорошим связям. Не последнюю роль играет и то, что соответствующие орбитали обычно высоки по энергии, и это также не самое лучшее условия для хорошего связывания. По этой причине простых комплексов, в которых были бы монодентатные лиганды Z-типа, маловато. Вот всего пара примеров, оба с производными алюминия в качетстве Z-лигандов. В одном мы без труда обнаружим в качестве такого лиганда хлористый алюминий, популярнейшую кислоту Льюиса из классической органической химии. Только в кординационной химии это нельзя так грубо и примитивно называть, а то он обидится и скажет: “Сами вы хлористый алюминий! Я трихлоралан, и это звучит гордо!” Да, все производные трёхвалентного алюминия надо называть по заместительной номенклатуре аланами, от родоначального алана AlH3, гидрида алюминия. И тогда сразу и отпадёт охота надсмехаться над, например, вот таким комплексом, рассаказывая, как некий незадачливый комплекс нульвалентной платины где-то подхватил молекулу хлористого алюминия, типа, колбу, наверное плохо помыли после реакции Фриделя-Крафтса. Нет, всё серьёзно, и мы видим, как комплекс Pt(0) с объёмистыми сильнодонорными фосфинами захватывает в координационную сферу трихлоралан (структура LIMGAQ из H.Braunschweig, K.Gruss, K.Radacki, Angew.Chem.,Int.Ed. 2007, 46, 7782).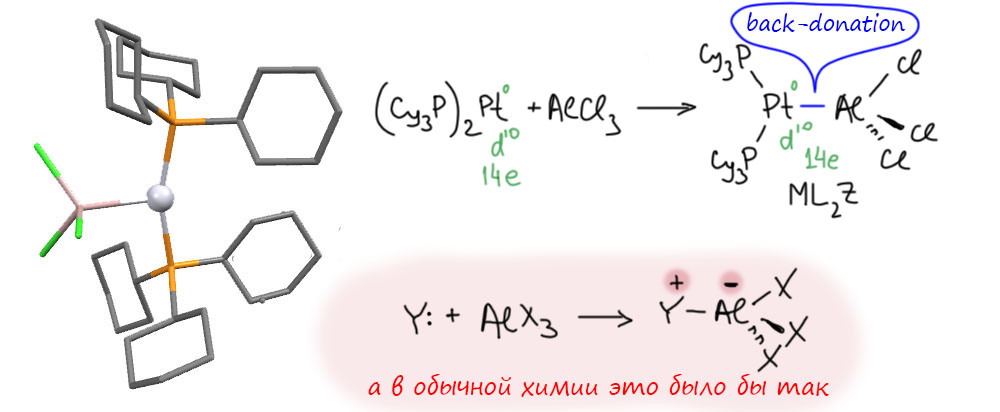
Заодно уточним, что лиганды Z-типа не изменяют ни счёт электронов, ни степень окисления переходного металла. И со скрипом сердечным поймём ещё одну премерзкую вещь – нам придётся отказаться от нормального отношения к донорно-акцепторному взаимодействию и со стороны непереходного металла. Ведь если бы у нас на трихлоралан напал бы нормальный нуклеофил (основание Льюиса), то мы бы получили тетраэдрический алюминий с зарядом -1, и назвали бы это аланатом (или алюминатом). Но как только у нас переходный металл, который взаимодействует как нуклеофил с помощью back-donation, мы получаем точно такой же тетраэдрический алюминий, но минус на нём не рисуем просто потому что тогда пришлось бы нарисовать плюс на платине, а мы условились так не делать, когда в ходу back-donation как основной (или дополнительный механизм связывания). Мы в упор не видим back-donation во всех формальных способах обозначения взаимодействия – в степенях окисления, счёте электронов, даже формальных зарядах. Такова неписанная конвенция, и на то она и неписанная, чтобы не носится с ней как с писаной торбой и не упрекать всех подряд, включая незадачливых деятелей ИЮПАК, что вот, взаимодействие есть, связь есть, а как она отражается на структуре приходится описывать только словами – типа, не дело, надо было бы изобрести какой-нибудь формальный способ. А мы знаем про ИЮПАК то, что там никогда не берут на себя смелость что-то изобретать новое, там только могут одобрить что-то, что и без них уже устоялось в химическом сообществе. А если в химическом сообществе ничего пока не устоялось, то и ИЮПАК не при делах. Впрочем, может быть, такая чрезвычайная робость химических законодателей и к лучшему, а то мы знаем, как в некоторых иных краях ретивость законодателей каждый день приводит к бессмысленным нововведениям и запретам.
Так получилось именно из-за переменного характера back-donation как механизма связывания, потребовавшего бы для учета введения дробных степеней окисления и прочих характеристик, а это противоречит духу и букве химии. Вот так и будем жить, и это нередко будет ставить нас в тупик в тех случаях, когда back-donation проявляет себя не как вспомогательный, а как важный или даже основной механизм связывания.
И еще одну любопытную вещь отметим. Z-лиганд неменяет счета электронов, но место в координационной сфере занимает, и следовательно всё же есть один формальный параметр, в котором этот лиганд учитывается – координационное число. Получается, что у Z-лигандов может быть одно любопытное свойство – они занимают координационное место в координационно-ненасыщенном лиганде, держат это место для каких-то будущих надобностей. Поскольку они довольно слабо связаны, заместить их в координационной сфере может нормальный лиганд достаточно легко.
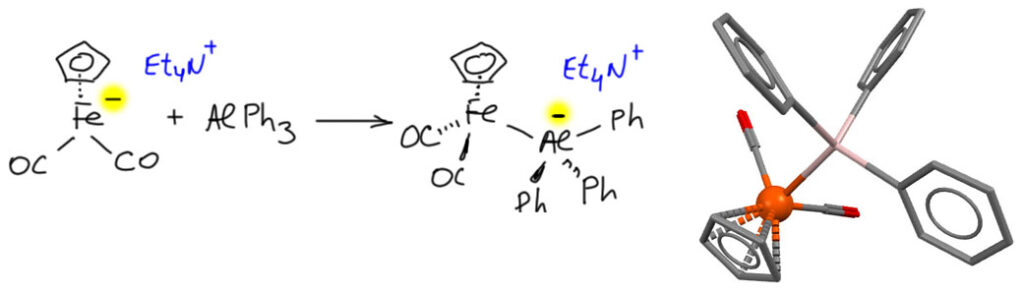
Вот ещё один пример, и тоже с соединением алюминия, трифенилаланом. В данном случае переходный металл реагирует в составе анионного комплекса, и мы вынуждены обозначить заряд, поместив его по всем правилам обычной химии на алюминии. Если бы мы этого не сделали, мы бы имели проблемы с сохранением заряда, а это очень тяжкий и непростительный грех. Но оцените финт – состояние алюминия совершенно одинаково в этом комплексе и в предыдущем. Но здесь мы пишем минус, а там нет. Будьте бдительны – на такой ерунде многие делают ошибки, и после долго ломают голову, куда делись или откуда взялись какие-то дурацкие заряды. И ещё один поучительный момент – Z-лиганд может быть весьма объёмистым и занимать немало места в координационной сфере, а такие вещи в химии с участием комплексов переходных металлов иногда бывают очень полезны. (комплекс PCFEAL взят из J.M.Burlitch, M.E.Leonowicz, R.B.Petersen, R.E.Hughes, Inorg.Chem. 1979, 18, 1097)
Но всё же большинство известных комплексов с Z-лигандами относятся к хелатам, причём Z-лигандный центр занимает одно место, а все остальные – обычные L или X-лиганды. Хелатирование облегчает связывание даже слабого координационного центра. Вот пример с бидентатным хелатом, Z-центр в котором – боран, довольно непростой, скорее даже это гетероциклическое соединение, борный аналог флуорена. Этот комплекс забавен не только своим потешным внешним видом, но и тем, что у металла все три координационных центра разных типов (комплекс XENXOE из S.Bontemps, G.Bouhadir, K.Miqueu, D.Bourissou, J.Am.Chem.Soc. 2006, 128, 12056)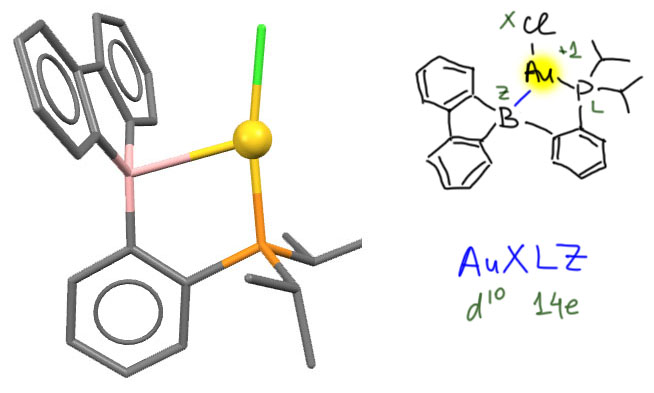
С лигандами Z-типа мы будем встречаться нечасто, но это интересные штуки, и рано или поздно мы к ним придём.