Связи металл-металл
Один из способов достижения координационной насыщенности комплексов металлов – образование димеров, ну и так далее – тримеров, тетрамеров, олигомеров, полимеров. Чаще всего это достигается с помощью мостиковых лигандов, но есть и возможность образования связей металл-металл. Начнём с этим разбираться, пока избежав разговора о кратных связях, ограничимся одинарными. И здесь нас поджидает несколько сюрпризов, например, окажется, что димеризация и олигомеризация карбонильных комплексов поздних переходных металлов умеет очень затейливым образом создавать видимость наличия связией металл-металл, на самом деле избегая их с помошью необычного вида мостиков. Попробуем разобраться в двух знаменитых молекулах – димерных карбонилах железа и кобальта, в случае успеха это откроет нам секрет построения очень большого количества карбонильных кластеров металлов 8-й и 9-й групп.
Кроме этого попробуем впервые сунуться в 12-ю группу: оказывается, там есть очень интересные структуры, которые в очередной раз напомнят нам о том, что металлы этой группы не совсем потеряли связь с миром переходных металлов. И в конце посмотрим на ещё один занятный тип гетерометальных комплексов, в которых один металл модет попробовать стать лигандом для другого.
Та система структур комплексов, которую мы тут усердно до этого момента разбирали, основана на одном предположении – металл всегда окружен лигандами, и даже если в комплексе несколько металлов, сборку их в комплекс мы осуществляем за счет мостиковых лигандов, которые легко анализируются по общим законам, и каждый атом металла в таком комплексе имеет свое собственное лигандное окружение. Фактически такой комплекс это просто несколько комплексов, соединённых мостиками, и это довольно типичная организация больших молекул самых разных типов, включая органические. И даже не только молекул, а и кристаллов – упаковка производных металлов в кристалл часто происходит за счет образования полимеров за счет сцепления отдельных одноядерных комплексом мостиками – это часто является проблемой, потому что такие кристаллы-комплексы часто плохо растворимы, ведь растворение для них возможно только за счет реакции расщепления мостиков, а это требует реакционноспособных молекул в реакционной среде, чаще всего, кислот, а это может быть неприемлемо с точки зрения других реагентов. Такие полимерные комплексы в кристаллах часто образуют простые производные меди, и с этим очень долго были связаны огромные сложности в развитии химии медь-органических соединений и катализа производными меди. Палладий в простых комплексах (“солях” – так мы по привычке называем всякие хлориды, ацетаты и т.п.) тоже часто полимерен, и это тоже причина того, что, например, такая простая и доступная “соль” как хлорид палладия совсем непопулярен как исходное для палладиевых реакций – растворимость совсем никуда, а расщепить эти бесконечные мостики можно только очень активными лиганлами с кислотным содействием.
Но есть и другой способ, с помощью которого координационно-ненасыщенные комплексы решают проблему ненасыщенности – образование димеров с прямыми связями металл-металл. А почему обязательно димеров? Могут быть и более крупные образования – тримеры, тетрамеры, и т.п. – полиядерные комплексы или кластеры. Именно такие комплексы называют кластерами в полном смысле этого слова. Причина эта довольно очевидна. Если мы на секунду отвлечёмся от комплексов в общем, и подумаем о комплексах металлов в степени окисления ноль, то быстро поймём, что образование связей металл-металл в этом случае это очевидный способ образования собственно металла как вещества из атомов металла. Мы с этим столкнёмся очень быстро как только озаботимся такой проблемой, как стабилизация нульвалентного металла лигандами. Такие комплексы должны быть координационно насыщенными, иначе они неустойчивы и очень быстро выделяют металл. Образование связей металл-металл начинается по свободным координационным местам, и продолжается по мере диссоциации лиганда – частица растет и это во всех смысла кластер. Как только атомов налипает подобольше, возникает ситутация, когда атомы в середине уже совсем не имеют лигандов, лиганды сохраняются только на периферии частицы, образуя такой защитный слой, препятствующий дальнейшему росту частицы – так образуются наночастицы.
Иными словами, получается такая немного странная ситуация, что один комплекс металла, имеющий свободное координационное место, служит таким большим лигандом для другого ненасыщенного комплекса металла. Просто связи металл-металл поэтому это в общем то же самое, что обычные ковалентные связи между любыми другими элементами. Металл (со своей координационной сферой) для другого металла – просто Х-лиганд, но не вносящий вклада в степень окисления по причине равности степеней окисления (если металлы одинаковые). Если разные, то по идее надо взять электроотрицательности Аллена и обычным образом прикинуть степени окисления, но это довольно экзотический случай.
Простые связи металл-металл в комплексах переходных металлов
В химии комплексов со связями металл-металл особую историческую роль сыграли металлы 7-й группы и особенно рений. Первые комплексы с подьверждённой связью металл-металл – димерные карбонилы марганца и рения (Dahl, L. F.; Ishishi, E.; Rundle, R. E. Polynuclear Metal Carbonyls. 1. Structures of Mn2(CO)10 and Re2(CO)10. J. Chem. Phys. 1957, 26, 1750– 1751) Мономерные карбонилы Mn(CO)5 и такой же карбонил рения – нульвалентные металлы 7-й группы, конфигурация d7, пять карбонилов дали бы для мономеру 17 электронов – не хватает до 18. Выход найден в объединении усилий, фактически мономер это радикал, сдваивание радикалов дает обычную ковалентную связь и искомую пару электронов. Структура комплекса – два октаэдра в заторможенной конформации. В димере каждый металл имеет 18 электронов (FUZTOJ: M.R.Churchill, K.N.Amoh, H.J.Wasserman, Inorg.Chem. 1981, 20, 1609)
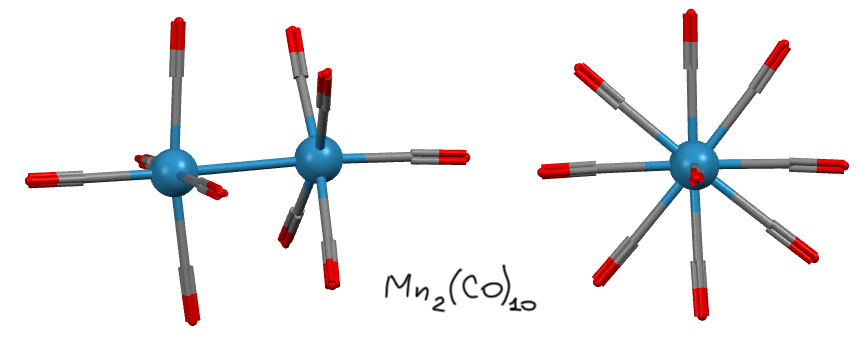
Похожая ситуация вроде бы с карбонилом металла 9-й группы, кобальта. Здесь мы бы имели Co(CO)4 – конфигурация d9, число электронов тоже 17, такая же история. Ожидаем тоже димера через связь кобальт-кобальт, например, вот такого, тоже в заторможенной конформации:
 А почему рисунок корявый, рентгена не нашлось что ли? Это же такое красивое кристаллическое вещество, кристаллики как рубины маленькие, неужели трудно РСА сделать. Конечно, полно есть структур карбонила кобальта, вот например, (FOHDEL01: P.C.Leung, P.Coppens, Acta Crystallographica,Section B: Structural Science, 1983, 39, 535). Что это? Как это устроено?
А почему рисунок корявый, рентгена не нашлось что ли? Это же такое красивое кристаллическое вещество, кристаллики как рубины маленькие, неужели трудно РСА сделать. Конечно, полно есть структур карбонила кобальта, вот например, (FOHDEL01: P.C.Leung, P.Coppens, Acta Crystallographica,Section B: Structural Science, 1983, 39, 535). Что это? Как это устроено?
Чтобы разобраться, как это устроено и в каком состоянии находится кобальт, разберёмся с похожей структурой, но немного более простой, потому что это хотя бы производное металла чётной группы, так что хотя бы нет проблем с неспаренными электронами. Это знаменитая структура, одна из тех, с которых вообще начиналось структурное изучение комплексов переходных металлов.
Так есть связь или нет?
В многоядерных карбонилах переходных металлов очень часто мы видим карбонильный лиганд, играющий роль мостика, связанного с двумя атомами металла через углерод Такой карбонил очень часто называют кетонным, намекая на сходство таких фрагментов со стуркрами кетонов. Совершенно очевидным следствием такого представления было бы то, что такой лиганд становился бы X-лигандом и изменял 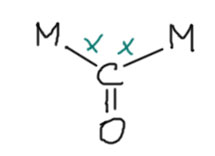 сепень окисления металла на единицу. И еще это должно было бы приводить к регибридизации углерода, сильному удлинению связи C-O по осношению в длине связи в обычных карбонилах, более напоминающей тройную. Так ли это?
сепень окисления металла на единицу. И еще это должно было бы приводить к регибридизации углерода, сильному удлинению связи C-O по осношению в длине связи в обычных карбонилах, более напоминающей тройную. Так ли это?
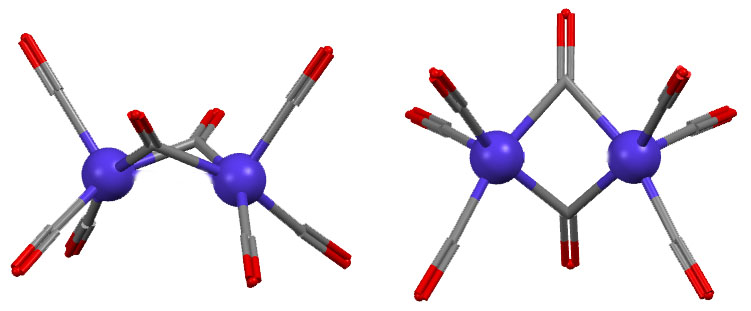
Скорее всего нет. Это одна из самых старых проблем в координационной химии переходных металлов. Самое типичное соединение с такой то ли фичей, то ли багом – нонакарбонил дижелеза (поклонников чистоты русского языка, в погонах или без, прошу расслабиться или почитать что-нибудь другое – нет ничего тупее борьбы за “чистоту языка”, особенно такого, который претендует на более широкое распространение и на роль в качестве языка общения разных этносов – живой и перспективный язык всегда развивается, заимствует слова, умные и глупые, пафосные и смешные, переваривает их и или выплевывает, или делает словами для словарей). Рентгеноструктурный анализ появился довольно давно, отец и сын Брэгги получили нобелевку за теоретические основы анализа рентгеновских дифрактограмм в 1915 году, и основали первый центр для реального применения этой науки, там все считали вручную, анализ самых простых структур занимал иногда годы, но качество работы было таково, что многие классические структуры, сделанные тогда, почти ничем не уступают тем, которые сделаны столет спустя на автоматических приборах с мощными компьютерами. Уже в 1920-х начинали делать структуры довольно сложных соединений, но обязательно тех, которые легко кристаллизуются и имеющих симметричные молекулы с тяжёлыми атомами, потому что дифрактограммы обсчитывали вручную и важно было, чтобы они были проще и контрастнее. Структура карбонила железа была одной из первых для этого метода (R. Brill, Z. Kristallogr., 1927, 65, 89–93). После структуру неоднократно переделывали, но ничего нового найдено не было. Наиболее качественная структура была сделана Коттоном и Траупом (F.A.Cotton, J.M.Troup, J.Chem.Soc.,Dalton Trans. 1974, 800 ). Вот она (код структуры FUZGAI).
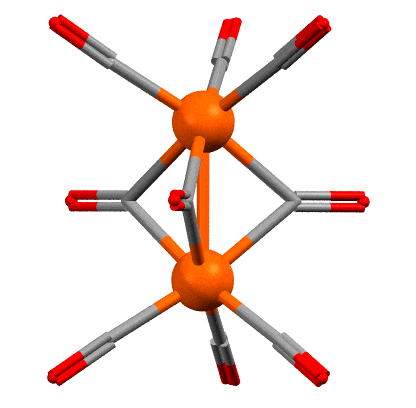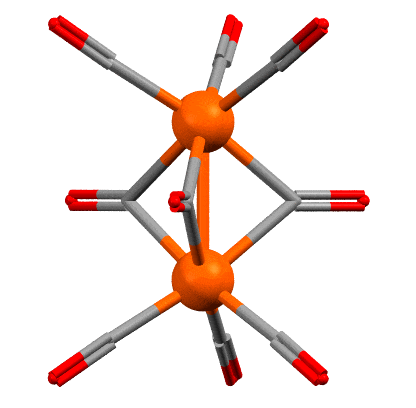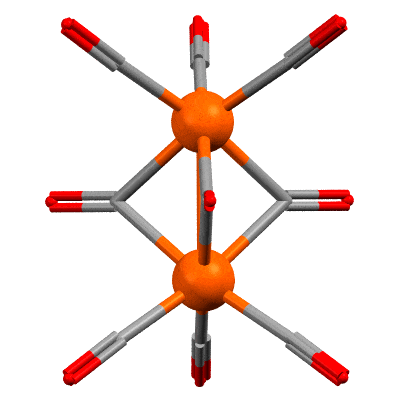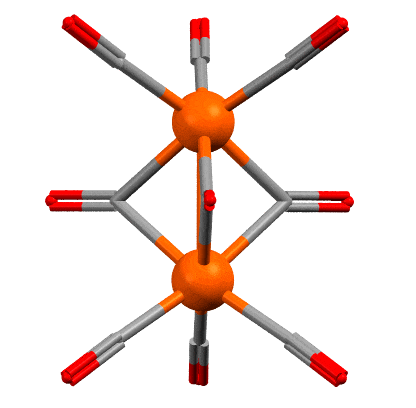
Между атомами железа очень часто рисуют связь железо-железо, вот как здесь. Но напомню, что рисовать не значит реально связывать – программы визуализации результатов РСА рисуют связи просто по критерию расстояния между атомами, устанавливая для каждой связи некоторый порог, ниже которого связь рисуется. Обычно это работает, но гарантий нет. Но среди структурных химиков понемногу, но давно сложился консенсус об отсутствии такой связи в этом соединении. Поэтому, лучше бы так (крутить уже не будем):
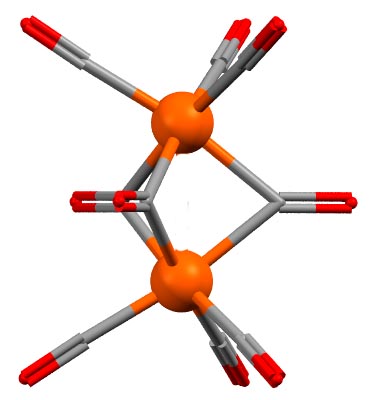
Наличие или отсутствие связи можно выяснить с помощью квантовохимических расчётов, причём не всегда это просто и могут потребоваться расчёты высокого уровня – здесть тоже не всё так просто, потмоу что в сложных многоатомных молекулах орбитали сильно делокализованы, а это значит что просто глазами – из разглядывания картинок молекулярных орбиталей установить наличие или остуствие связи между конкретными атомами почти невозможно. Как же оно тогда устроено, и как посчитать электроны в таком соединении, а это нелишне, потому что в химии карбонилов такая ситуация встречается нередко. Сам нонакарбонил дижелеза образуется весьма просто: при аккуратном фотолизе обычного мономерного карбонила, а для этого его достаточно выставить на солнце, происходит диссоциация одного карбонила и два фрагмента немедленно соединяются. Один из них координационно насыщенный, другой нет.
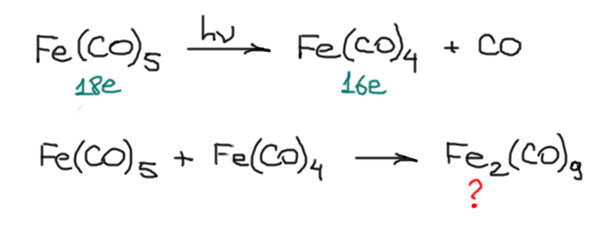
Ясно, что мы в очередной раз видим ситуацию, когда координационно-ненасыщенный комплекс (тетракарбонил железа) ищет, как бы насытить координационную сферу. И поглядев вокруг себя не видит ничего кроме еще неразложившегося насыщенного пентакарбонила. Кажется, это тупик – нет ничего, никаких лигандов безхозных. Придется попробовать уговорить пентакарбонил поделиться карбонильными лигандами компромиссным способом, так чтобы поделить пару электронов лиганда между двумя атомами железа. Это называется трёхцентровой связью. В принципе, если мы помним, как устроен диборан, мы поймём и это. В диборане есть координационно ненасыщенный или, как там это называют, секстетный атом бора, и ничего кроме гидрида. Вот на паре электронов от гидрида и двух пустых орбиталях двух атомов бора и создается трехцентровая связь. В диборане их две, эти связи слабее обычных двухцентровых, поэтому диборан более-менее легко диссоциирует, на чём и основана химия гидроборирования. Нечто подобное образуется и при связывании двух атомов металла через мостик карбонила, ориентированного неподелённой парой внутрь. Сама структура готового димера образована двумя железотрикарбонильными фрагментами, очень часто встречающимися в комплексах Fe(0). В “нормальных” комплексах такой фрагмент связывает два двухэлектронных или один 4-хэлектронный лиганд, например, диен. В этом случае эту роль играют три карбонила, то есть вроде бы 6 электронов, но эти электроны находятся в общей собственности трёх атомов. Никаких ценных указаний, как надо в этом случае считать электроны у атомов металла нет. Если поделить поровну, получим по 3 электрона, итого 17 на металл, но мы понимаем, что нечетное число вызывает ощущение того, что это система с открытой оболочкой, но это не так – важно то, что каждая трёхцентровая связь обслуживается парой, то есть оболочка закрыта. Поэтому, мы можем сойтись на этих 17 электронах, но с оговоркой, что это кажущееся, эффективное число. Здесь логика такая: не 16 электронов, потому что именно такая ненасыщенность заставила тетракарбонил железа вступать в эти связи. Не 18 электронов – на это просто не хватает.
Получается, что желание насытится в этом комплексе реализовано не полностью, и это очень хорошо, потому что делает этот комплекс более лабильным, чем исходный пентакарбонил. И это действительно тоже играет на сравнение с дибораном. Тетракарбонил железа совсем ненасыщен, поэтому не существует в устойчивом виде – ищет ещё один лиганд. Пентакарбонил железа наоборот слишком устойчив, и вводится в реакции как правило, в условиях фотолиза. А вот нонакарбонил дижелеза реагирует с другими лигандами практически спонтанно – видимо, благодаря спонтанной диссоциации по слабым трехцентровым связям. Это основное исходное для получения железотрикарбонильных комплексов.
Судя по структуре продукта, фрагменты становятся неразличимы, а молекула приобретает плоскость симметрии ровно посрединке между атомами железа, и в этой плоскости располагаются три общих карбонила. Очевидно, что таким образом каждый атом железа достигает координационной насыщенности. Но как? А может всё же не достигает. Как устроена структура димерного карбонила долго было неясно, и можно найти немало статей, в том числе самых крупных исследователей, где предлагаются варианты. В результате дискуссий некоторое подобие консенсуса было найдено, и проблему можно считать решенной. В этом месте надо только очень хорошо понимать, что конкретно является проблемой. Не существует никакой проблемы в том, чтобы сделать расчёт молекулярной и электронной структуры и этого соединения и любого другого устойчивого соединения в основном состоянии, и мы буез труда или с трудом получим, например, описания молекулярных орбиталей всей молекулы на таком высоком уровне теории, что ожидать каких либо улучшений к этой картине не придётся. Электронная структура даст нам распределение электронной плотности, заряды, порядки связей, и кучу всего ещё. Так что же нам нужно? Удобной интерпретации, котора позволит нам считать, что мы понимаем структуру – как она образовалась. Ничего удобнее, чем разбиение структуры на отдельные связи никто так и не придумал. удивительно, насколько легко большиснтво структур подчиняются разбиению на самые простоые связи между парой атомов, обслуживаемой парой электронов – двухцентровые двухэлектронные связи (2с-2уe). И кратные связи между элементами мы представляем несколькими простыми двухцентровыми. Немного сложнее со связями с гапто-лигандами – там точно не получится сводить к двухцентровым, но мы сохраняем двухэлектронность, разбивая связь с лигандом с гаптностью выше двух на несколько двухэлектронных, что и позволяет нам использовать формальный эквивалент гапто-лиганда (типа как Cp мы представляем как XL2). Эта схема отлично работает, хотя нам приходится придумывать, как на нее сверху наложить сильные смещения электронной плотности и в случае мезомерии, и в случае значительной доли участия back-donation, но ни там, ни там мы не трогаем основу системы – а самое главное, чо в ней есть это то, что мы всё время придерживаемся электронной пары, как такой неделимой единицы связывания. Неделимой не в том смысле, что её нельзя разделить, а в том, что если мы это сделаем, немедлено попадем в такой параллельный мир – мир молекул с неспаренными элетронами, open-shell, радикалами и прочей нечистью, чур-чур нас от этого – заметите ещё как тщательно я этого избегаю, и что это неплохо получается, а за бортом оказывается не так много всего интересного, хотя оказывается, и рано или поздно я сдамся и мы поедем и туда тоже. Но пока не поедем, мы ещё в мире closed-shell далеко не со всем разобрались.
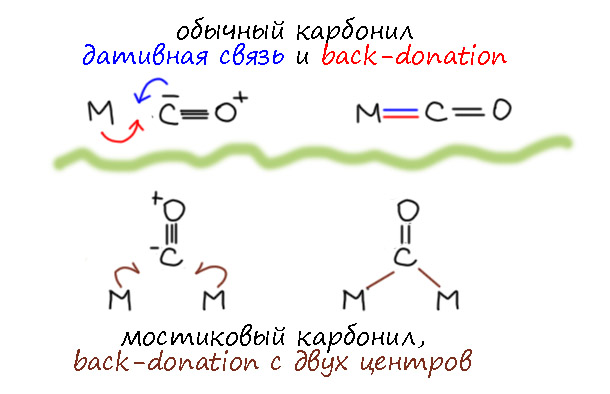
Связь металл-карбонил-металл принято считать трёхцентровой, но двухэлектронной. Иными словами, это такой случай, когда лиганд L-типа, карбонил, связан с двумя металлами Льюиса координационной связью, но одновременно. Английский исследователь координационной химии переходных металлов Малькольм Грин даже предложил для такой связи отдельный символ – одностороннюю стрелку (J. C. Green, M. L. H. Green, G. Parkin Chem. Commun., 2012, 48, 11481–11503), но на мой взгляд, это плохая идея, потому что с такими стрелками обычно связывают взаимодействия и смещения неспаренных электронов.
![]()
Здесь это создает сомнительное узнавание, потому что кажется, что это удачно говорит нам о том, что пара обслуживает два взаимодействия, типа, почему бы ее пополам не поделить, ну и стрелку тоже уполовинить. Но фокус в том, что пару нельзя половинить – это по-прежнему закрытая оболочка (это термин, соответствующий closed-shell). Само по себе взаимодействие легко представить на уровне комбинации орбиталей фрагментов, если мы используем на карбониле ту же самую sp-гибридную орбиталь, и подходящие d-орбитали на железах.
Вернёмся к карбонилу кобальта, который должен был быть димером со связью Co-Co, но в рентгене почему-то оказался вот таким: (FOHDEL01: P.C.Leung, P.Coppens, Acta Crystallographica,Section B: Structural Science, 1983, 39, 535). Что это? Как это устроено?
Теперь, зная как устроен димерный карбонил железа, мы узнаём и устройство кобальтового. Естественно, поскольку у кобальта на один электрон больше, то и на один карбонил меньше. Получается, что димеризация двух мономерных тетракарбонилов кобальта может происходит за счёт двух трёхцентровых связей через карбонилы. В этом месте у нас некоторое удивление – а неспаренные электроны? Ведь трехцентровые связи сделаны за счет вакантных ордиталей на металла и пары на карбониле, они не затрагивают неспаренный электрон. Во-первых, для нас, органиков, это шок – как можно использовать для связи что-то другое, когда у вас фактически свободные радикалы! Они же сдваиваются всегда и везде, если только этому не помешает стерика. Но здесь она точно не мешает, потому что “нормальный” димерный карбонил точно существует и содержится в растворах в равновесии с этим. Но этот карбонил, с мостиками, устойчивее. Судя по всему, ненамного, на 1-2 ккал/моль, но два мостика стабильнее прямой связи Co-Co. Вполне можно поверить, потому что связи металл-металл обычно очень слабые. Ладно, смиримся, а что всё-таки с неспаренными электронами.
Что точно и известно сразу про неспаренные электроны выражается хорошо известной, но несколько потерявшей остроту формулой: их там нет. Молекулы карбонила хрома диамагнитны и не проявляют никаких признаков наличия неспаренных электронов. Но может быть, это синглетный бирадикал, бывают такие – два отдельных неспаренных электрона, но с противоположными спинами. Хорошо, но если эти электроны находятся на металле, то неизбежно их взаимодействие в пространстве между атомами металла, а такого взаимодействия не видит ни один из способов обнаружения ковалентного связывания (в квантовой химии таких несколько, и их применяли и к карбонилу железа, и к карбонилу кобальта, но не нашли). И вот ведь какая фигня: всё же не забудем, что в растворе (по ИК-спектрам) фиксируется и димер по связи металл-металл, а значит, скорее всего, и мономер, хотя мы этого, насколько я знаю, не знаем, и не исключено, что изомеризация двух димерных молекул происходит без диссоциации, а просто за счет движения карбонильных лигандов.
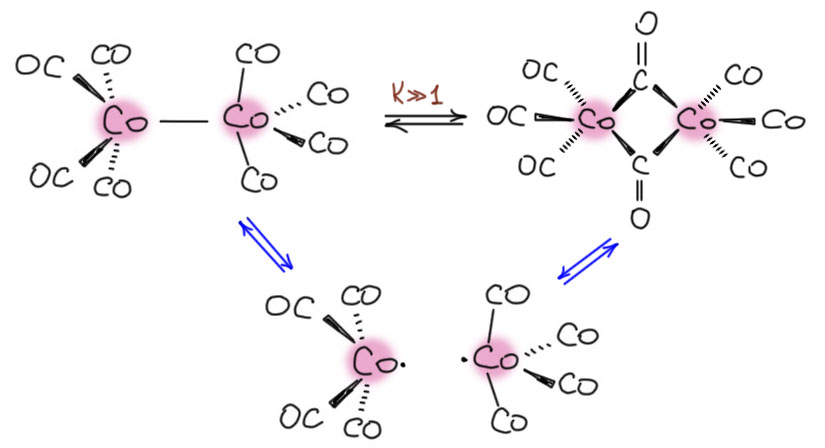
Более устойчивый димер с мостиковыми карбонилами безусловно устроен так же как и димерный карбонил железа, но тут уже просто так невозможно притянуть трёхцентовую связь и считать, что на этом тема закрыта, потому что надо ещё объяснить, куда подевались неспаренные электроны – такое впечатление, что они умудряются как-то спариться при образовании этих мостиков. Но как? Может быть эти две вещи связаны? Неужели этим никто не занимался? Занимались конечно. Вот уже полвека приблизительно каждые 10 лет выходит новая теоретическая статья, применяющая новый теоретический подход для объяснения структуры карбонила кобальта. Даже удивительно, зачем так много, ведь если какая-то из статей была убедительна, то вопрос должен был быть закрыт. Но нет, не закрыт, хотя бы потому, что подходы каждый раз особые, нестандартные, и внятной картины связывания не дают – вас просто предлагают поверить в очередную теоретическую концепцию. И каждый раз это оказывается не очень убедительно, и требует новой концепции.
Я не буду всё это разбирать, потому что дело, на мой взгляд, не так сложно, и все можно объяснить без дополнительных концепций. Зададим себе просто вопрос – куда могли подеваться валентные электроны с металла, и не видели ли мы такое уже в других случаях. К сожалению, если мы попали сюда, только начиная наше путешествие по комплексам переходных металлов, то пока что мы немного видели. Но скоро увидим, как элегантно и эффективно снимают электроны с металло лиганды, способные на сильный back-donation – такие лиганды ещё называют пи-кислотами, потому что за приём электронов с металла отвечают орбитали пи-типа, ещё точнее, вакантные орбитали пи-типа, а это всегда одна из нижних незанятых орбиталей (НСМО или одна из ближайших следующих), по типу они всегда строго разрыхляющие (строго говоря, в сложных молекулах различие между связывающими и разрыхляющими орбиталями не совсем корректно, если не иметь в виду самую нижнюю поносимметричную, и противостоящую ей в заоблазных высях виртуальную полнонесимметричную; лучше говорить о занятых и незанятых или виртуальных). А давайте посмотрим повинимательнее сначала на геометрию этого димера. Я его посчитал 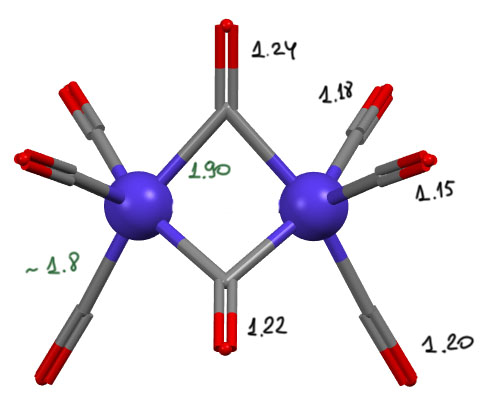 обычным DFT (B3LYP, полноэлектронный базис jorgedz), получилось очень близко к экспериментальной геометрии в кристалле, хотя дины связй в мостике чуть-чуть занижены. Вот данные рентгена. Опа! Посмотрите-ка, расстояния C-O довольно сильно различаются, и в мостиковом карбониле они совсем длинные – это уже почти двойная связь C=O! Напомню, что расстояние C-O выдаёт порядок связи; в карбониле, присоединенном только координационной связью, это практически чисто тройная связь, а это 1.12-1.14 А. А если на карбонил идёт электронная плотность с металла по эффекту back-donation, связь разрыхляется (потому что плотность идёт на разрыхляющую пи-орбиталь карбонила), и становится ближе к двойной – чем больше плотности туда влито, тем длиннее связь, тем ближе к двойной. И ещё мы вспоминаем, что back-donation, увы, эффект переменный, смещение плотности обычно частично, и разрыхление пи-связи неполное, мы по удлинению может судить о величине эффекта. И вот мы видим здесь, что именно на мостиках эффект максимален, а на обычных карбонилах он меньше, причем те карбонилы, которые висят в транс-положении к мостикам тоже сильно разрыхлены, а те, что под углом (можно назвать это цис- по отношению к мостикам), намного короче, хотя всё равно слегка удлинены. И что это значит? Как минимум то, что карбонилы в карбониле кобальта работают как пи-кислотные лиганды, причём мостиковые карбонилы в большей степени.
обычным DFT (B3LYP, полноэлектронный базис jorgedz), получилось очень близко к экспериментальной геометрии в кристалле, хотя дины связй в мостике чуть-чуть занижены. Вот данные рентгена. Опа! Посмотрите-ка, расстояния C-O довольно сильно различаются, и в мостиковом карбониле они совсем длинные – это уже почти двойная связь C=O! Напомню, что расстояние C-O выдаёт порядок связи; в карбониле, присоединенном только координационной связью, это практически чисто тройная связь, а это 1.12-1.14 А. А если на карбонил идёт электронная плотность с металла по эффекту back-donation, связь разрыхляется (потому что плотность идёт на разрыхляющую пи-орбиталь карбонила), и становится ближе к двойной – чем больше плотности туда влито, тем длиннее связь, тем ближе к двойной. И ещё мы вспоминаем, что back-donation, увы, эффект переменный, смещение плотности обычно частично, и разрыхление пи-связи неполное, мы по удлинению может судить о величине эффекта. И вот мы видим здесь, что именно на мостиках эффект максимален, а на обычных карбонилах он меньше, причем те карбонилы, которые висят в транс-положении к мостикам тоже сильно разрыхлены, а те, что под углом (можно назвать это цис- по отношению к мостикам), намного короче, хотя всё равно слегка удлинены. И что это значит? Как минимум то, что карбонилы в карбониле кобальта работают как пи-кислотные лиганды, причём мостиковые карбонилы в большей степени.
В тех случаях, когда мы подозреваем сильный эффект back-donation, крайне полезно прикинуть, что было бы, если бы эффект развился на полную катушку, как полноценная связь, с полным смещением плотности. В этом случае это тоже полезно, и есть еще одна тонкость – карбонил висит на двух металлах, и поскольку молекула симметрична, плотность будет снимать с обоих. Если мы более-менее разобрались, как устроены обычные карбонильные лиганды – оксид углерода образует координационную связь с металлом, используя свою пару (а точнее сигма-связывающую ВЗМО – ее долю со стороны атома углерода) и дополнительно возникает back-donation, в зависимости от валентного состояния металла более или менее сильное, иногда это смещение плотности от металла к углероду карбонила на пи-разрыхляющую НСМО можно считать второй свяью, пи-связью, но рисуют ее отдельной связевой чертой кране редко именно потому что эффект имеет переменную величину, и каждый раз надо было бы как-то судить связь это уже, или просто электронный эффект. А теперь возьмём карбонил и два металла в конфигурации мостика, то есть два металла и углерод образуют треугольник, а кислород направлен вовне – как могло бы осуществиться такое связывание? С одной стороны, вполне можно представить координационную связь за счет перекрывания одной из вакантных d-орбиталей (только вот есть ли такие у нульвалентного кобальта, имеющего почти полный комплект d-электронов, так что вакансий у Co(0)не видно, разве что s и p-АО, но такое перекрывание вряд ли будет эффективным, и электроны останутся почти целиком на лиганде. А вот наоборот очень легко себе представить связывание – back-donation, но сразу от двух металлов. Вот такая связь, назовём это связью, но не будем упираться в вопрос, а не изменилась ли степень окисления кобальтов, – не принято так делать, по крайней мере, в химии карбонильного лиганда. И отсюда же видно, куда могли подеваться неспаренные электроны с атомов кобальта – они попали в одну область связывания, а значит успешно спарились просто по определению, так как поселились в одной области пространства, И как некоторый побочный результат от обсуждения гипотезы о том, что неспаренный электрон тоже отлично может участвовать в back-donation, становится приблизительно ясно, почему такой слабой оказывается связь Co-Co в димере без мостика – эффект back-donation обусловливает делокализацию неспаренного электрона (коротко это называют спином – делокализацию спина) и на металле остаётся небольшая спиновая плотность, недостаточная для эффективного взаимодействия и образования прочной ковалентной связи. Более выгодной поэтому и оказывается ситуация, когда за димер отвечают мостики.
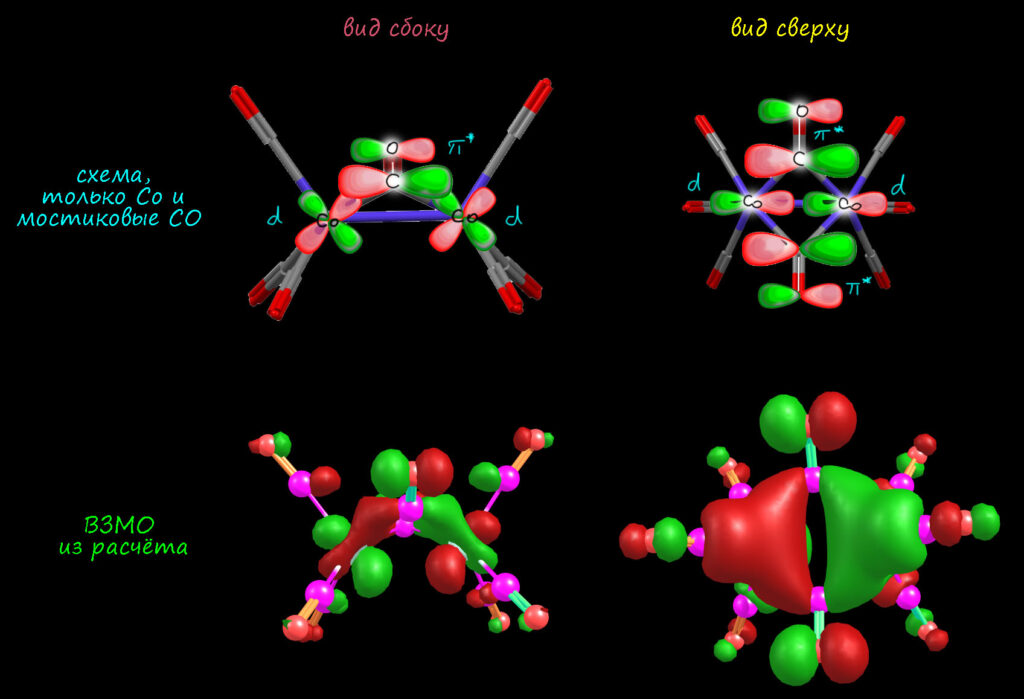
И чтобы получить некоторое подтверждение такой возможности, поразглядываем молекулярные орбитали димерного карбонила кобальта. Достаточно посмотреть на самые верхние заполненные, начиная с ВЗМО. Логика здесь проста: димеризация это при прочих равных самое слабое взаимодействие, димер слаб, а за взаимодействия молекул с наименьшей энергией (или по-другому, за наибоее выраженные типы реакционной способности) отвечают как раз самые доступные, высшие занятые орбитали. При этом мы не настолько наивны, чтобы ожидать, что в молекулярных орбиталях прямо и найдём те самые трёхцентровые связи: метод МО почти никогда ничего не говорит о тех связях (двух или трёхцентровых), с которыми привык работать стуктурный или органический химик; молекулярные орбитали почти всегда сильно делокализованы и включают вклады от всех атомных ориталей (или орбиталей фрагментов), которые могут перекрываться в одних областях пространства, приблизително подходят по энергии и симметрии. Связь мы не найдём, но можем увидеть интересующее нас взаимодействие и понять, что это скорее – back-donation или обычная координационная связь. Это легко понять, если мы в молекулярных орбиталях увидим части орбиталей отдельных карбонилов. Хорошо, смотрим на ВЗМО (расчёт мой). Я спецтально привожу два вида – сбоку и сверху – на МО, из расчёта (внизу), а выше схематически прикидывают, как они получились из атомной орбитали металла и орбитали карбонила. Я соблюдаю области перекрывания (одим цветом – связывание: два цвета с узлом-промежутком между – антисвязывание). И еще я для простоты и наглядности убрал из схемы все карбонилы кроме мостиков, потму что мы сразу видим по расчетному ВЗМО, что действительно, наибольший вклад дают именно мостики и это именно антисвязывающие пи-орбитали, которые отвечают за пи-кислотность и прием плотности с металла по back-donation. И мы видим, что у металла работает одна из d-орбиталей (какая именно неважно, для этого нужно добавить оси координат, но это для нас здесь лишняя информация) и что взаимодействие этой орбитали и пи-орбиталей карбонилов именно связывающее, одними долями (одним цветом, одним знаком, одной фазой, как хотите). Удивительно, но картинка ВЗМО прямо показывает, что это ровно то, что мы хотели найти, даже как-то неудобно, но нет-нет, я всё честно нарисовал и ничего не подгонял. За взаимодейсвие фрагментов в димере действительно отвечает back-donation с металла на москтиковые карбонилы, именно поэтому они удлинены больше других. Но это не полная ковалентная связь. Откуда я знаю? Из той же геометрии – если бы взаимодействие зашло далеко до высокой степени ковалентности, мы бы увидели регибридизацию углерода в sp2 и угол Co-C-Co поближе в 120°, но в реальной структуре угол даже немного острый (среди других МО есть и другие подтверждения, но нам хватит этих).
Вот такая забавная картина того, что может быть в некоторых димерных карбонилах, а значит и в тримерных и олигомерных – в химии карбонилов переходных металлов особенно поздних переходных металлов много таких структур. Мы увидели, что в образовании таких мостиков очень большую роль играет back-donation (можем смело обобщить под моё честное слово – я разглядывал и другие примеры и неизменно наверху были орбитали, соответсвующие back-donation). А что тогда со счётом электронов? Увы, никакой протсой схемы здесь нет, а сложная схема для такой простой и наглядной вещи как счёт электронов просто не имеет смысла. Собственно, мы можем точно скзать, что такие димеры и олигомеры говорят о координационной ненасыщенности, поэтому такие карбонилы всгда имеют большую реакционную способность, чем мономерные 18е-карбонилы (в тех случаях, когда такие существуют, например, у железа, но не у кобальта или родия), а для образующихся из них комплексов ообычно можно проще прикинуть число электронов и всё остальное, что например, и происходит в знаменитой реакции Посона-Кханда (до неё мы ещё доберёмся).
12-я группа просится в контекст
Cо связями металл-металл мы можем легко доехать до 12-й группы, которая у нас висит в воздухе и которой мы отказываем в праве называться переходными металлами. Но ведь именно там мы впервые сталкиваемся со связью металл-металл, в соединениях одновалентной ртути, а такая вещь как каломель Hg2Cl2 это вообще нечто древнее, не совсем-совсем древнее, скорее старинное – в 18-19 веках это было лекарство против всего, типа, всё равно помирать, но с каломелью веселее. Покопавшись в памяти наших детских воспоминаний о химии, мы, пожалуй, поймём что это не только первое, но единственное соединение из простой химии, в котором можно заподозрить прямую связь между атомами металла. И у нас нет в этом сомнений, при том, что димерных формул мы видели немало. Например, хлорид одновалентной меди мы записываем точно так же как каломель – Cu2Cl2, но мало кому прийдёт в голову, что в этом соединении есть связь медь-медь. Степень окисления металлов одинакова, и разница в том, чётное или нечётное число валентных электронов. У Cu(1+) оно чётное, конфигурация d10, у ртути нечётное, конфигурация d10s1 – фактически это радикал, а радикалы обычно рекомбинируют. А меди приходится использовать мостиковые лиганда для димеризации. Конечно, и мы даже не удивлены, ведь в ртути есть что-то, на выбор: мистическое, таинственное, неметаллическое, подозрительное, зловещее. Сейчас-то нас ничем не удивишь, а когда-то давно люди сильно удивлялись тому, насколько прочные связи с углеродом образует ртуть, совсем не так как другие металлы (в старину в переходные совсем не лезли и под металлами понимали всякие натрии-магнии, в самом крайнем случае алюминии). Поэтому то, что ртуть может образовывать прямо самые настоящие радикалы, которые рекомбинируют в димеры, и не вызывало диссонанса.
В 12-й группе, раз мы в неё залезли, связи металл-металл для степени окисления +1 очень долго были известны только для ртути. Причина этого та же самая, что и в других хорошо известных особых свойствах этого металла – он очень хорошо держит свои валентные электроны на валентной s-оболочке. В приниципе, мы согли бы особо не париться, и просто сослаться на хорошо известные нам тенденции в изменении свойств переходных металлов в ряду, что собственно и дает нам полезную классификацию на ранние и поздние, и аккуратно, оглядываясь по сторонам, шёпотом сопоставлять поздние с неметаллами. И приписать ртуть к поздним переходным металлам. И сделать вид, что мы не утверждали, что группа цинка особая, и что причислять их к переходным нельзя, потому что валентная оболочка у них не использует d-электроны. И ведь это чистая правда, d-оболочка закрыта, ушла вглубь (в смысле энергии, но и размера тоже, в результате обычного сжатия). Это как раз и является причиной того, что элементы этой группы ведут себя как активные металлы, напоминая металлы второй группы – d-оболочка эффективно экранирует валентную s-оболочку от ядра, и способствует тому, что электроны из нее очень легко уходят. Только мы должны сделать оговорку – в этом описании мы легко узнаём цинк, но не другие металлы группы, которые намного более прижимисты и электронами так легко не разбрасываются. Уже кадмий намного более инертный металл, не зря кадмий-органику обычно получают не прямым внедрением металла в связь углерод-галоген, как мы легко делаем с цинком, а переметаллрованием из магнийорганики. Ртуть же совсем уже напоминает скорее неметаллы. В последние десятилетия это стали объяснять релятивистскими эффектами – огромный заряд ядра и очень сильное сжатие электронных оболочек приводят к тому, что скорость движения электронов становится очень высока. В этом месте многие удивятся – что-то мы не очень часто рассуждаем о скорости электронов, мы ведь привыкли считать их такими облаками, окутывающими ядра – а что эти облака там двигаются? И стоя на ядре наблюдатель сможет сказать – сегодня что-то очень пасмурно, электронные облака закрыли небо, скоро пойдёт дождь. Хи-хи-хи (это ехидный смех, а не три греческие буквы χ), но облака облаками, но ведь революцию в теории атома совершила модель Нильса Бора, и никто с тех пор не говорил, что это плохая модель. Отличная модель, с тех пор она просто обросла математикой, но актуальности не потеряла. А в этой модели электроны именно движутся по орбитам, и идея Бора состояла просто в том, что орбиты не произвольны, а подчиняются строгим законам и кроме того дискретны, квантованы. Дальше из этого сделали красивую теорию с волновыми функциями, как источником всей информации о состоянях электрона, но опять таки, никто не отменял главного – законы квантования орбит оперируют таким вполне механическим понятием как момент количества движения – это идет ровно с той модели Бора, в которой у электронов есть орбита, и движение по ней описывается в общих чертах так же как движение спутников вокруг планеты, только с добавленем квантования. Чем больше притяжение и уже орбита, тем быстрее надо крутиться. Вокруг атома ртути надо крутиться просто с бешеной скоростью. Единственное, во что здесь придется поверить, потому что проверить это можно только усердно въехав в математику, это то, что учет релятивистских эффектов ведет к увеличению энергии связи электронов с ядром. С эти действительно связано очень многое не только у ртути, но и вообще у всех элементов нижних периодов Системы.
Поэтому ртуть такая. Поэтому она легко отбирает электроны у тех, кому он не так нужны – с эти, в частности, связано образование амальгам, чрезвычайно экзотермическое для электроположительных металлов – например, щелочных, или алюминия. В структурах амальгам щелочных металлов просто видны катионы щелочных металлов и олигомерные анионы ртути (со связями Hg-Hg, более слабыми чем в каломели, потому что это электроноизбыточные кластеры). Поэтому ртуть предпочитает не ионные, а ковалентные связи даже с неметаллами, что, среди прочего и дает нам ртутьорганические соединения, весьма легко образующиеся и очень прочные. И даже между атомами ртути мы видим ковалентные связи.
Для кадмия и особенно цинка это несвойственно. Тем не менее некоторая общность свойств есть, и соединеня со связями металл-металл все же получили и для кадмия, и для цинка. Первым таким соединением цинка стал димерный комплекс с пентаметилциклопентадиеном (Cp*Zn-ZnCp*). Прежде чем разобраться с ним, посмотрим вообще как обстоят дела у цинка с циклопентадиенильными комплексами, цинкоценами. Они давно получены и с разным замещенными циклопентадиенилами. И ни один не устроен как настоящий сандвич с двумя пентагапто-лигандами. Все устроены как то, что называют сдвинутым сэндвичем, один лиганд пентагапто, второй моногапто. Вот так, хотя бывают небольшие вариации в пространственном расположении моногапто-кольца.
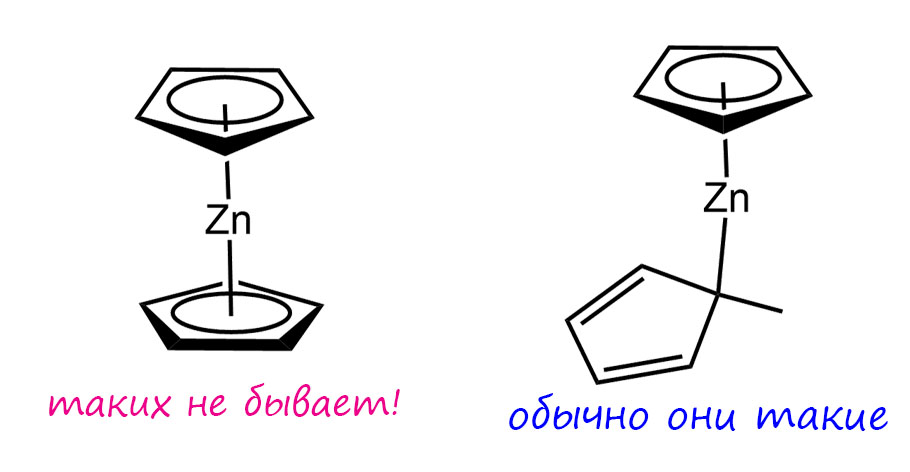
Посчитаем электроны, хоть цинк и не переходный металл. Но d-оболочка есть, в Zn(2+) она полная, d10. В настоящем сэндвиче было бы 10+6+6 = 22. Ничего себе! Слава богам, что цинк не переходный металл. А во второй? Ой, 10 +6 +2 = 18. Идеально! Получается, что цинк тоже подчиняется 18-электронному правилу и это и объясняет, почему структура такая. Вот так открытие!! Я думаю, что это дрянное открытие, смысла в нём ноль. Ведь если мы всё же примем, что у элементов группы цинка d-оболочка заполнена, погружена вглубь, неактивна в валентном смысле, то и считать эдектроны там глупо. Покойтесь с миром, d-электроны! Мы не потревожим ваш покой. И если считать валентной оболочкой только s и p, то за вычетом 10 мы получаем 8 – обычный октет! Цинкоцен со сдвинутым лигандом подчиняется правилу октета. Из этого, в частности, сдедует весьма волнительное умозаключение, что цинк (и его коллеги по группе) очень старается следовать тем закономерностям связывания лигандов, которые работают у настоящих переходных металлов – циклопентадиен может быть пентагапто-лигандом с 6-ю электронами, отдаваемыми металлу по обычным механизмам координационной связи. Но – не используя d-электроны, только с возможностями, обычными для элементов непереходных, элементов главных подгрупп, p-элементов. А можно связать пентагапто-циклопентадиенил только с помощью sp-оболочки? Да без проблем. Трём занятым π-МО циклопентадиенильной системы аккуратно по симметрии соотвествуют три p-АО, а полносимметричной ещё и s-АО – связывай не хочу, это и даёт одну σ- и две π-связи.
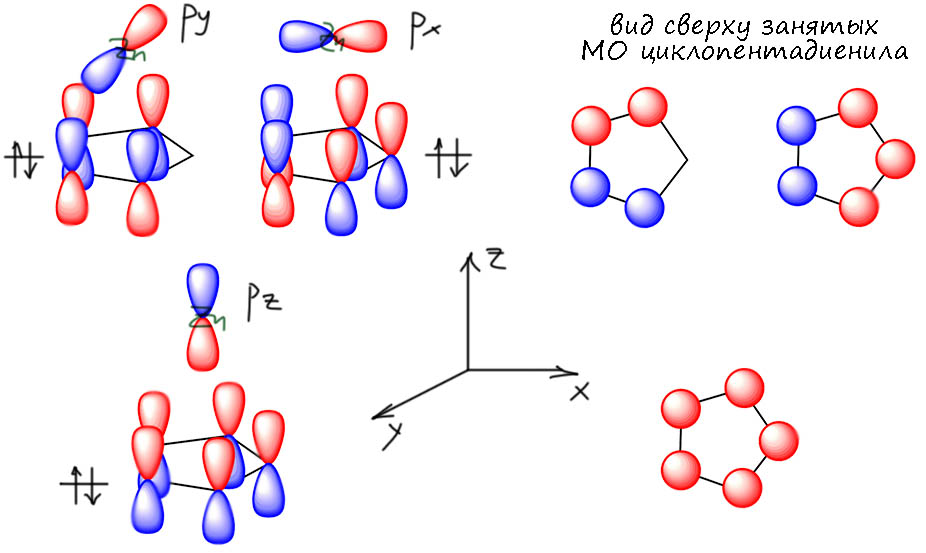 Ну а второй Cp связан простой связью как обычный X-лиганд. Для этого у цинка осталась ещё s-орбиталь. И всё нормально – устройство цинкоцена становится очевидным.
Ну а второй Cp связан простой связью как обычный X-лиганд. Для этого у цинка осталась ещё s-орбиталь. И всё нормально – устройство цинкоцена становится очевидным.
Здесь уже возникает другая проблема. В главной подгруппе старой второй группы, той, в которой находилась и подгруппа цинка, находится бериллий, магний и щёлочноземельные металлы. И у всех есть металлоцены, причем бис- и сэндвичевые, со структурами, издали вообще неотличимыми от ферроцена. А это ведь всё s-элементы, даже не p, к тому же там точно нет даже внутренней d-оболочки. Но 18-электронный счет для бериллия и щелочноземельных металлов тоже смысла не имеет, даже сильно формального. Что же получается – цинк не может взять два пентагапто-Cp, только один, а вся эта убогая братия s-элементов из второй группы, о которой мы редко вспоминаем иначе как просто про катионы в солях, имеет больше возможностей, чем цинк, металл всё же серьёзный, последний из d-блока, у которого точно по крайней мере хотя бы p-оболочка уже опустилась в валентную зону без вопросов.
А вот скорее всего нет. Скорее всего, все эти от бериллоцена до бароцена металлоцены – склеены ионными связями с минимальной примесью ковалентности. Cовсем отрицать возможность координационной связи на s-АО этих металлов было бы странно, хотя и их свойства (они так же чувствительны к влаге и воздуху как CpNa), и структура (расстояния от металла до колец очень хорошо описываются именно ионными радиусами, а сами кольца немного елозят и скособочиваются на шарике иона металла, что невольно выдает ненаправленность взаимодействия). И обратно сравнивая эти ионные металлоцены с цинкоценам, который тоже мог бы использовать ионные связи, но тогда у него не было бы проблем и с вторым кольцом – а раз он предпочитает уважать октет, значит взаимодействия всё же координационные, а не ионные.
Вернёмся к цинкоцену. В 2004-м Эрнесто Кармона и его коллеги из Севильи и Мадрида аккуратно изучили реакцию между цинкоценом (не c обычным Cp, а с намного более донорным Cp*) и диэтилцинком – статья получилась аж в Science (I.Resa, E.Carmona, E.Gutierrez-Puebla, A.Monge, Science 2004, 305, 1136). То, что должно было быть обычной реакцией обмена, лигандов привело к необычному результату – фактически редокс-реакции, и образовался до тех пор невиданный Zn(1+) в виде димера со связью Zn-Zn. Очень трудно понять, почему сигма-связь цинк-углерод в моногапто-циклопентадиенильном комплексе вдруг так легко принимает лишний электрон, что приводит к обрзованию цинкового радикала и сдваиванию. Факт в том, что это не единичный пример такой реакции, и позже было найдено что точно так же реагирует и гидрид – как одноэлектронный восстановитель. А впрочем… давайте подумаем, что точно произошло в этой реакции. Был один Cp в состоянии просто одновалентного заместителя. То есть не ароматического. Перенос электрона на связь цинка с этим лигандом делает и этот Cp полноценным циклопентадиенильным ароматическим пента-гапто лигандом, который охотно подхватывает катион этилцинка, образовавшийся при одноэлектронном окислении диэтилцинка. Можно представить, что переход от моно-гапто в пента-гапто довольно выгоден – и вот это и есть причина этой реакции – ароматизация всех циклопентадиенилов и образование еще одного комплекса с цинком. Мы уже пришли в тому, что связь металла с пента-гапто-Cp это фактически тройная связь. В этом месте, конечно, мы можем вспомнить из обычной органической химии, что тройная связь прочнее одинарной, но она же вносит большую нестабильность. Но в применении к комплексам с циклопентадиенилами это наверняка не так – в этом случае нет большой концентрации электронной плотности в малом пространстве между двумя атомами, так как атомов здесь все шесть. На этом и остановимся – понятна нам стала движущая сила этой не очень обычной реакции. И понятно почему то же самое происходит при реакции с другими легкоокисляющимися анионами. И ещё один довод – если мы примем, что простая связь на моногапто-Cp и правда обслуживает s-орбиталь цинка, то мы не можем не вспомнить, что s-орбиталее более компактные чем p-орбитали, и лучше удерживают электроны – а эта тенденция только усиливается в периодах от 3-го и ниже – и тогда нас тем более перестаёт удивлять то, что обычное замещение вдруг приобрело редокс-характер.
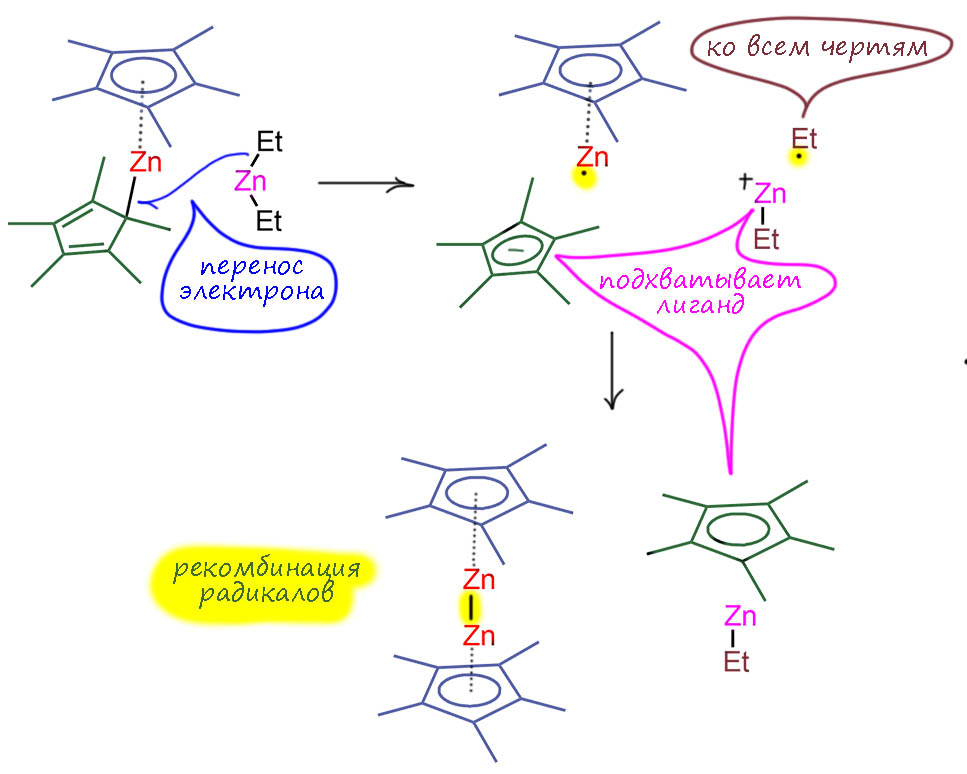
И вот как устроена эта молекула (код LACTUF: I.Resa, E.Carmona, E.Gutierrez-Puebla, A.Monge, Science 2004, 305, 1136) – хороший сэндвич, только вместо одного металла пара, поэтому расстояние между блинами большое, и конфигурация заслонённая, а не заторможенная, как у обычных металлоценов. И поскольку связь цинк-цинк обслуживантся парой, у цинка здесь полный октет точно так же как у полусдвинутого цинкоцена. После открытия этого первого преставителя соединений цинка со связью металл-металл, такие производные стали обнаруживать и исследовать. 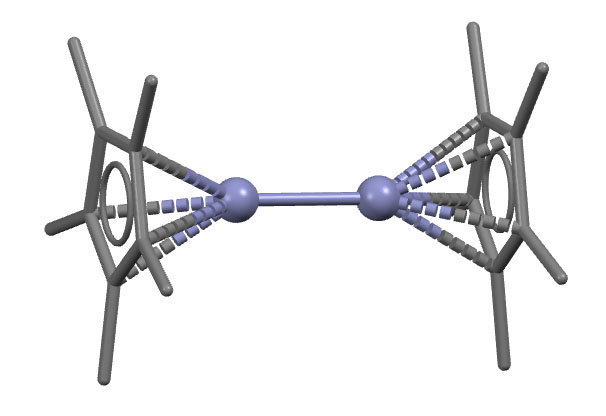
Надо сказать, что у цинка много применений в органической химии, и много интересных механизмов, так что обнаружение возможности образования промежуточной степени окисления и соотсветствующих комплексов может дать интересные идеи. Мы пока эту тему оставим, но не так уж и исключено, что когда-нибудь вернёмся. Мы решили не трогать эту группу, но зарок не давали, и не преминем обратить внимание на что-то интересное.
Металл металлу лиганд
Мы договорились не рассматривать связи между переходными металлами в двух и многоядерных комплексах как отношение металл-лиганд. Хотя могли бы считать один металл X-лигандом относительно другого, но нам чрезвычайно трудно было бы понять, кто из них кто. Да, если металлы разные, можно было бы взять электротрицательности по Аллену и формально разделить связь, считая менее электроотрицательный атом металлом, а другой – лигандом. Хорошо, а если металлы одинаковые, а это очень частый случай? Причем комплексы с одинаковыми металлами и с разными могут быть просто идентичны по структуре. Нет, это плохая идея, поэтому мы рассматриваем многоядерные комплексы именно как многоядерные, у каждого металла своя координационная сфера, и эти сферы и сами металлы как-то взаимодействуют.
Если металл непереходный, мы уже увидели, что можно ввести ещё один тип лигандов – Z-лиганды (чёрт побери, и что теперь с этим делать? – оставим, лиганды не виноваты, что где-то массово впали в трудное детство, а система обозначений международная, вменяемые люди не обязаны корректировать свою жизнь по чьим-то диким фантазиям). Z-лиганды мы рассмотрели отдельно, эта категория понемногу завоевывает признание, и скоро мы увидим такие комплексы в деле.
А если металл переходный? Можно всё же найти примеры, когда один переходный металл со своей координационной сферой работает как Z-лиганд по отношению к другому переходному металлу. А как тогда будет работать второй металл по отношению к первому? Видимо, как L-лиганд? Ой, как интересно! А что, такое бывает??
Ещё как бывает. Комплексов таких немало. Не то, чтобы много, но и немало – сотни точно, и в некоторых областях их очень любят, потому что это довольно интересный способ построения нновых архитектур больших многометальных систем. Но мы туда пока не полезем, а рассмотрим самое простое – два металла. Первые комплексы такого типа описал, видимо, все тот же Коттон, с которым мы скоро будем разбирать кратные связи металл-металл. Принцип очень элегантный, извините за это малость снобское словечко но тут оно уместно – что элегантно, то элегантно. Элегантность тут понадобилась потому что связи такие всё же большая редкость, и чтобы найти структурные мотивы, которые им благоприятствуют, пришлось серьёзно призадуматься, да сделать много экспериментов. Металлы не любят даже намёка на такие связи по понятной причине – очень много электронов на двух атомах рядом, отталкивание, Паули – враг кратных и даже простых связей между тяжёлыми элементами. Пришлось подобрать такие фрагменты, которые как-то дополнительно такие связи подпирают. Обычно это мостиковые лиганды, но Коттон с сотрудниками нашёл примеры более простых комплексов, но тоже с изюминкой.
Задача такая. С одной стороны нам нужен металл с сильной возможностью к back-donation. Значит что-то позднее, из последних групп. Второе – долен быть лёгкий подход к этому металлу, чтобы не протискиваться между лигандов – связи будут слабые и протискиваться никто не захочет. Ага, плоскоквадратная конфигурация – сверху и снизу дорога открыта. Значит d8 и 10-я группа, палладий или платина – лучше металл побольше. Теперь хорошо бы, чтобы у него была нуклеофильность побольше (мы знаем, что у переходных металлов нуклеофильность и back-donation это очень близкие вещи, одно обеспечивает другое). Вот тогда бы анионный комплекс взять, а не нейтральный. А второй металл должен иметь заметную электрофильность (быть кислотой Льюиса), и тоже поменьше лигандов. Здесь выбор мог бы быть побольше, но есть очевидный первый кандидат – 11-я группа, опять пониже, серебро, золото. Почему не медь? Медь очень любит образовывать агрегаты, многоядерные комплексы, которые не захотят разрушаться из-за какой-то слабой связи. А вот серебро и золото очень легко образуют весьма координационно ненасыщенные комплексы только с одним лигандом. В общем виде получается как-то так.
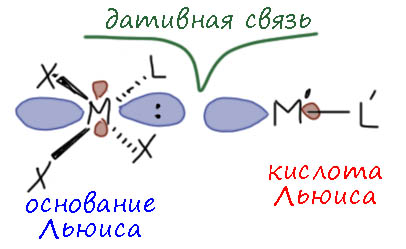
Это весьма интересная перспектива. Если это произойдёт, то мы получим двухядерный комплекс, который быдет комплексом с каждой из сторон. Это будет комплексом металла 10-й группы (палладия, платины) в степени окисления 2+, с конфигурацией d8 и координационным числом 5 – тетрагональная пирамида, в апикальном положении второй металл. Этот новый лиганд будет Z-типа, связанный только за счет back-donation. Такой лиганд не даёт вклад ни в счет электронов (по прежнему 16), ни в степень окисления. Получается небольшой, но яркий парадокс – явным желанием этого металла было насытить координационную сферу. Лиганд добавил, а мечту не выполнил. Из этого прямо следует такой забавный вывод, что есть ещё к чему стремиться, и сзади есть ещё местечко. Но мы пока не знаем, можно ли реально этим воспользоваться. Оставим интригу. И одновременно это комплекс второго металла, 11-й группы, в степени окисления +1 и с конфигурацией d10. До образования новой связи это был сильно координационно ненасыщенный комплекс с счётом электронов всего 12. Для него тот первый металл со своим лигандным окружением – настоящий лиганд L-типа (дал пару электронов на вакантную орбиталь, образовав дативную σ-связь). И счет электронов увеличился до 14 – получился довольно типичный для 11-й группы линейный 14-тиэлектронный комплекс, всё ещё ненасыщенный, но благодаря специфике 11 группы, не очень сильно желающий дальше расширять координационную сферу.
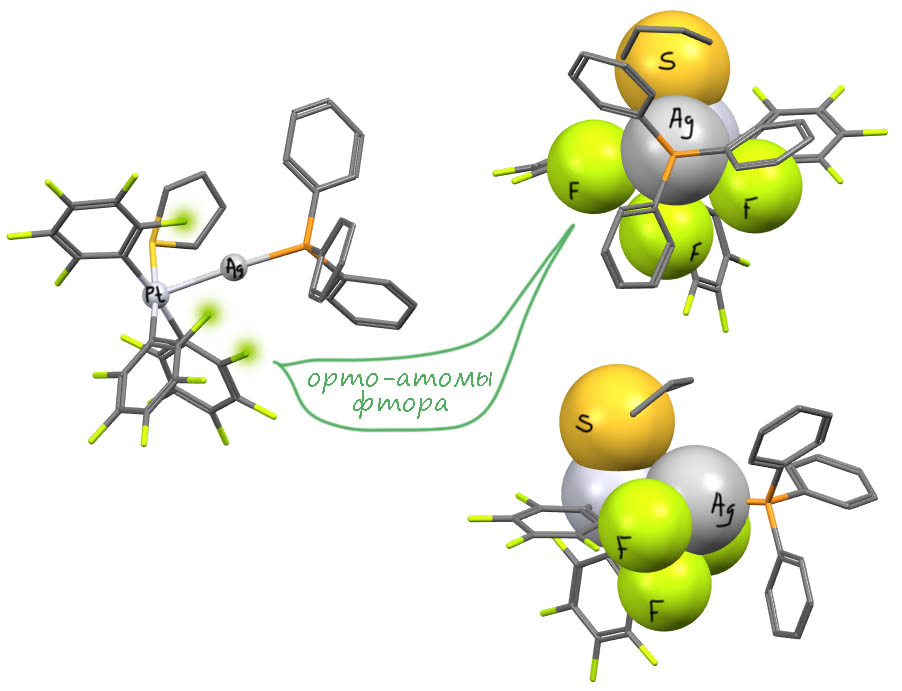
Посмотрим на реальный пример такого комплекса из статьи Коттона и сотрудников (F.A.Cotton, L.R.Falvello, R.Uson, J.Fornies, M.Tomas, J.M.Casas, I.Ara, Inorg. Chem., 1987, 26, 1366). Взаимодействует анионный комплекс платины с тремя пентафторфенильными лигандами и одним тетрагидротиофеном и катионный комплекс серебра с трифенилфосфином. Комплексы весьма слабые, хранятся только при -20°С, при комнатной температуре разлагаются хоть в твёрдом состоянии, хоть в растворе. Но минимум с одного удалось сделать РСА, и мы его сейчас посмотрим.
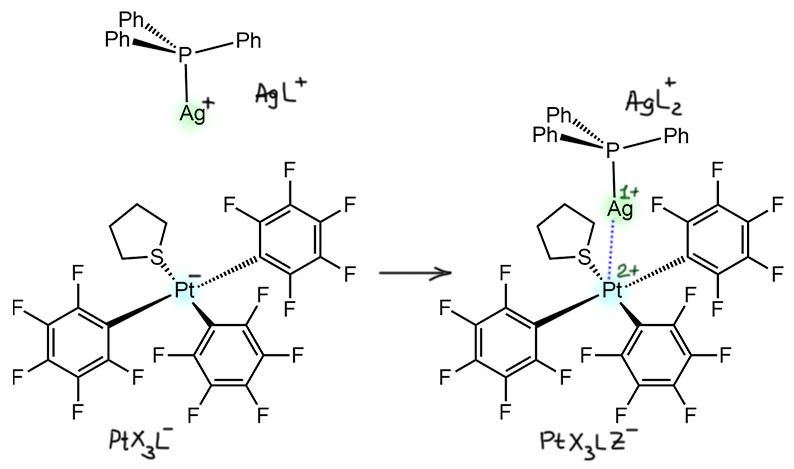
Обратим внимние на одну проблему. Оба исходных комплекса заряжены, и следовательно продукт не имеет заряда. А куда делись заряды? Если посмотреть со стороны серебра, то платиновый комплекс со своей парой и зарядом с виду неотличим от анионного лиганда. А почему это тогда не то же самое, что взаимодействие с любым другим анионным лигандом, да хоть хлоридом? Тогда мы получаем незаряженный комплекс серебра, но и лиганд в этом случае это X-лиганд.
Увы, этот подход немедленно даст парадокс. Потому что если с точки зрения серебра это X-лиганд, значит и с точки зрения платины тоже. Но мы, во-первых ничего не знаем про квадратно-пирамидальные комплексы платины, и во вторых это получится PtX4L с Pt(4+) – получается, что кто-то успел окислить платину. Кто такой шустрый, и кто тогда восстановился? Нет такого? Значит, это чепуха. И нам не надо переосмысливать тип лигандов, Z у платины, L – у серебра, а это значит, что формальные заряды никуда не делись со своих металлов, ну а в сумме это ноль. В некотором смысле это напоминает старую добрую семиполярную связь, но это тоже ложный след. Придется признать, что это очень особенный тип связи между переходными металлами.
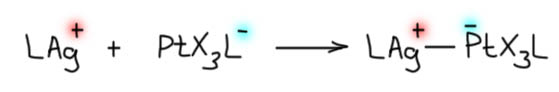
Рассмотрим структуру (код CUXWAT10: F.A.Cotton, L.R.Falvello, R.Uson, J.Fornies, M.Tomas, J.M.Casas, I.Ara, Inorg. Chem., 1987, 26, 1366). В ней есть ещё одна очень важная особенность – перфторфенильные лиганды на платине. Сам Коттон обратил внимание на важность именно таких лигандов (перхлорфенильные тоже годятся). Оказалось, что они играют очень важную роль в том, чтобы эта очень слабая свзь хоть немного дополнительно укрепилась. Смотрим сначала на саму структуру – всё, как ожидается, платина в плоском квадрате, ставшем основнием пирамиды, в вершине которой серебро. Связь длинная (2.6 Å), но, поверим Коттону, самая короткая из всех до того известных, в любом случае, она существенно короче суммы ковалентных радиусов, что точно свидтельствует о ковалентности. То, что она при этом слаьая, мы знаем по реальным свойства этого комплекса. Но вот что мы видим – серебро окружено со всех сторон атомами фтора и атомом серы – они прямо обнимают серебро. Я прямо показал эти атомы ван-дер-ваальсовыми радиусами, чтобы было понятно, как они сближены. Что это значит – дополнительное связывание, хотя бы электростатическое? Очень может быть. Но, на мой взгляд, даже скорее то, что они экранируют серебро от взаимодействия с внешними лигандами, а именно это и растаскивает связи – лигандный обмен с более сильными лигандами. Нет лигандного обмена, и комплекс более-менее стабилен. Вот зачем эти перфторфенильные лиганды на платине. Без таких комплексы этого типа получать не удавалось.