
Винилиденовые комплексы переходных металлов
Как образуются винилиденовые комплексы
Как мы уже выяснили, винилиден находится в очень близком родстве с ацетиленидным комплексом. Основной путь возникновения винилиденовых комплексов в реакциях – атака электрофила на β-атом в ацетилениде. Электрофилом может быть протон из какой-нибудь слабой протонной кислоты, имеющейся в реакционной смеси.

Обратим внимание на то, что металл не изменяет степени окисления, а значит при переходе от ацетиленида (X-лиганда) к винилидену (L-лиганду) на металле появляется формальный заряд, соответствующий степени окисления.
Довольно часто, особенно с комплексами рутения, винилиденовый комплекс образуется сразу при взаимодействии с терминальным ацетиленом, например,

Из этого примера видно, что винилиден является L-лигандом, и общий счет электронов сохраняется. И также видно, что вторая связь между металлом и углеродом рисуется просто для красоты и чтобы подчеркнуть карбеновую природу этого лиганда, и она никак не влияет ни на счет электронов, ни на степень окисления.
При взаимодействии такого типа водород как будто перескакивает с терминального положения на второй атом углерода, поэтому этот процесс иногда называют 1,2-миграцией. Но не стоит вкладывать в это слово лишний смысл – типа, водород согласованно перемещается как это бывает в сигматропных перегруппировках в органической химии. Вполне возможно, это происходит просто, так как протоны обычно и перемещаются между разными атомами – в реакционной смеси есть что-то основное, например, растворитель. Тогда протон переносится просто от кислоты к основанию и обратно, и так по цепочке перемещается между атомами.
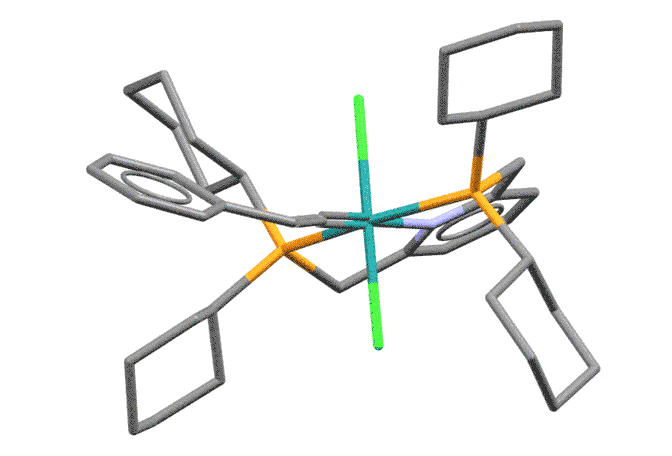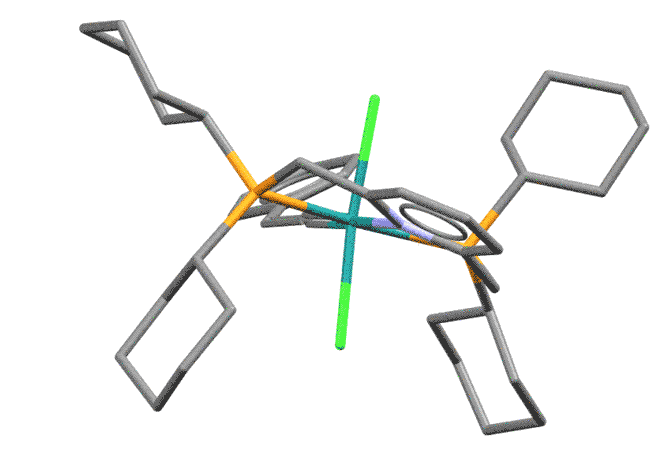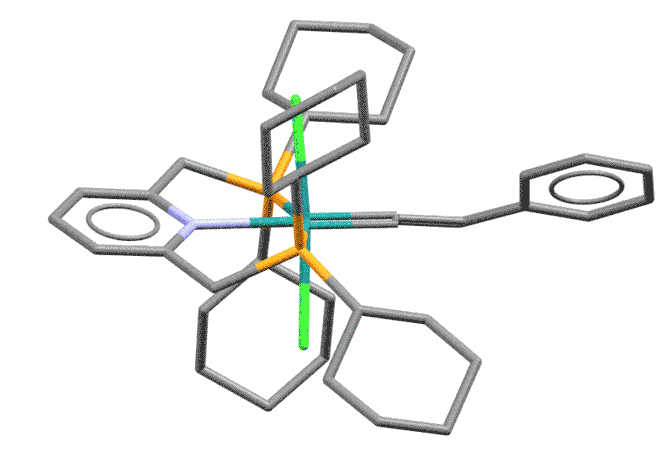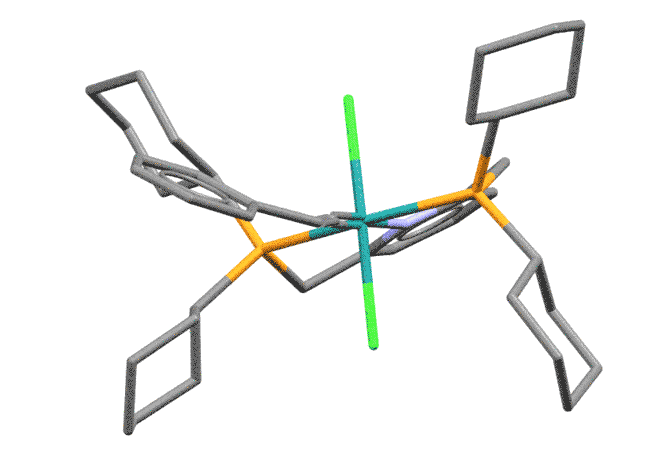
Вот несколько структур таких комплексов для примера:
Cp(CO)2Mn(=C=CMe2) (H.Berke et al, J.Organomet.Chem. (1981), 218, 193)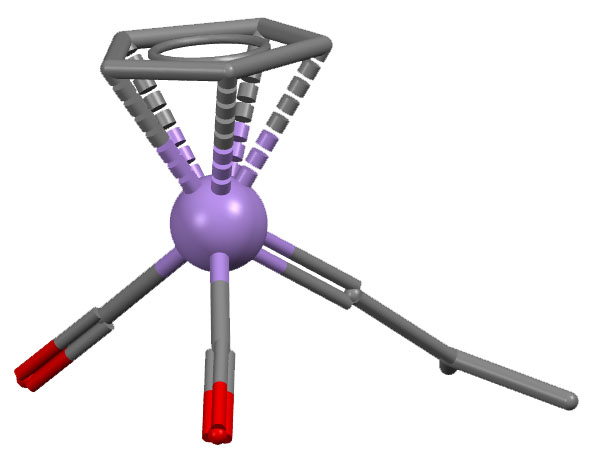
Cp(P(OMe)3)2Mo(=C=CHPh)Br (R.G.Beevor et al, Chem.Commun. (1983), 673)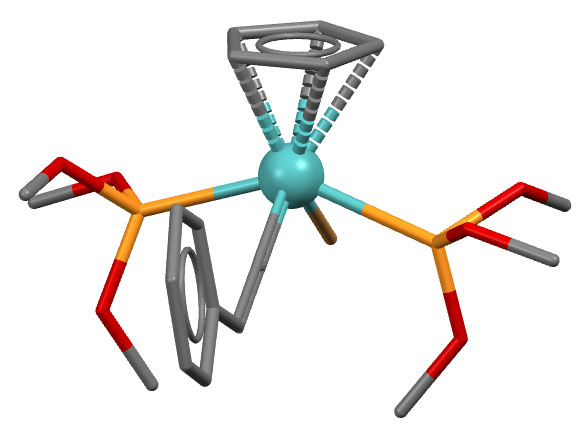
Cp(P(iPr)3)Rh(=C=CHPh) (J.Wolf et al, Angew.Chem.,Int.Ed. (1983), 22, 414)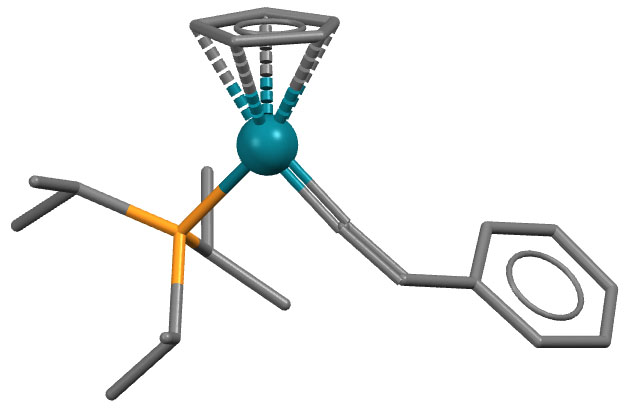
Комплекс рутения с хелатным дифосфином пинцерного типа (H.Katayama et al, Organometallics (2002), 21, 3285)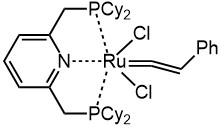
Транс-гидроборирование: родий-катализируемая реакция
Гидроборирование двойных и тройных связей, хоть обычное, хоть каталитическое всегда дает продукт син-присоединения. Для алкинов это значит, что заместитель из ацетилена и бор оказываются по разные стороны двойной связи. Когда полученное борорганическое соединение попадает в кросс-сочетание, которое идет с сохранением конфигурации, получаются транс-замещенные алкены. А как получить цис-замещенные? Здорово было бы иметь еще и другую реакцию гидроборирования – с анти-присоединением. Такая реакция долго оставалась мечтой, но в 2000 все тот же Мияура с сотр. нашел такую реакцию (JACS, 2000, 122, 4990).
Оказалось, что комплексы родия(1+) или иридия(1+) катализируют транс-гидроборирование терминальных алкинов, в присутствии донорных триалкилфосфинов и обязательно триэтиламина. Оба металла ведут себя почти одинаково, но родий чуть-чуть лучше, поэтому на нем и остановились. В конце концов после всяких оптимизаций остановились на таком протоколе (на схеме забыл указать триэтиламин!).
![]()
Во-первых, видим, что реакция, увы, не слишком селективная. Образуются всегда оба изомера, и в некоторых случаях преимущество цис-изомера совершенно мизерное. Более-менее неплохо получается с фенилацетиленами. Во-вторых, присутствие триэтиламина обязательно. Считается, и даже доказано, что если триэтиламина нет, то комплекс Rh(1+) с донорным фосфином прежде всего присоединяет боран, после чего реакция идет по обычному механизму, который мы уже разбирали: окислительное присоединение – вход ацетилена – миграционное внедрение – восстановительное элиминирование, что и приводит к обычному транс-изомеру продукта гидроборирования. Триэтиламин вызывает обратную к окислительному присоединению реакцию.
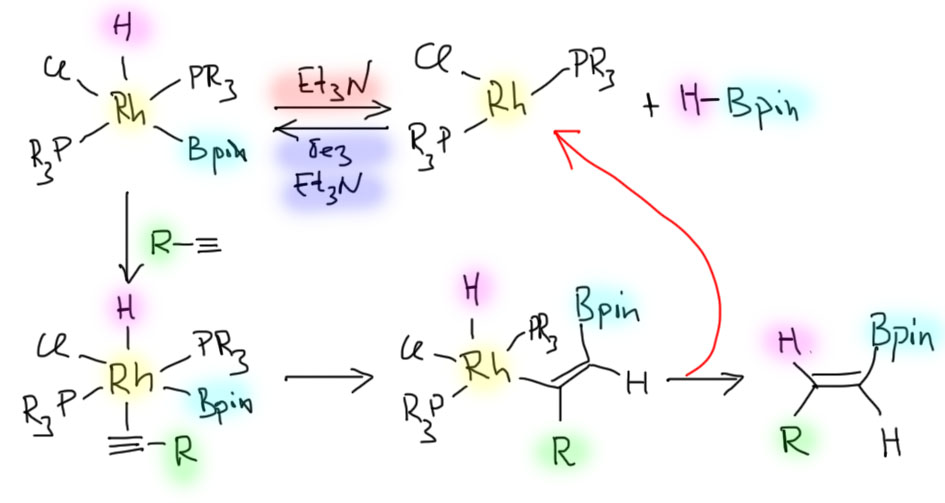
Механизм необычного транс-гидроборирования включает образование винилиденового комплекса. Как происходит превращение η2-ацетиленового лиганда в винилиденовый мы уже обсуждали – здесь важно то, что протон с терминального ацетиленового углерода переезжает на второй атом, и это подтверждается дейтериевой меткой. В работе (Fernandez et al, Chem. Eur. J., 2012, 18, 1512), где этот процесс еще раз хорошенько изучили, еще и предполагают, что это происходит через окислительное присоединение C-H связи к родию(1+), то есть вводится еще один промежуточный комплекс Rh(3+), который переходит в винилиденовый с уменьшением степени окисления, но эта гипотеза немного вилами на воде писана*, и мы ее опускаем. Оставим в этой части цикла везде Rh(1+). Далее мы окислительно присоединяем боран, тут же борильный остаток претерпевает миграционное внедрение на карбен винилиден, и реакция счастливо завершается восстановительным элиминированием продукта. Причина стереоселективности – стерическая: возможность получить огромный родий со всей своей лигандной сбруей и бывший заместитель из ацетилена по одну сторону двойной связи не очень привлекательна, поэтому они стараются расположиться по разные стороны, подальше друг от друга. Борильный заместитель, даже с рогатой пинаконовой нахлобучкой все же гораздо скромнее родия по размеру. Обращаю внимание на часто встречающееся недоразумение – это транс-гидроборирование, хотя получается цис-изомер продукта – стереохимия присоединения определяется относительным расположением фрагментов того, что присоединяется – здесь это борный остаток и водород.
Итак, цикл включает две степени окисления родия и включает вполне стандартные элементарные стадии входа лигандов, окислительного присоединения, миграционного внедрения и восстановительного элиминирования. Высокая реакционная способность низковалентного родия поддерживается донорными триалкилфосфиновыми лигандами.
![]()
* общепринятый краткий синоним словосочетания “подтверждена квантовохимическими расчетами пути реакции по методу функционала плотности“
Транс-гидроборирование: катализ комплексом рутения
Другая каталитическая система, дающая такой же результат в гидроборировании, основана на очень занятном комплексе рутения (Leitner et al, JACS, 2012, 134, 14349). Разберем этот кейс, он дает возможность чуть лучше понять, как работают винилиденовые комплексы. Исходный комплекс рутения весьма непрост. Это комплекс Ru(2+) с октаэдрической геометрией. Из 6 мест три жестко занимает тридентатный лиганд, в котором два фосфина и один пиридиновый азот. Этот лиганд остается на своем месте как вкопанный, оставляя на всю каталитическую химию три свободных места. Вначале их занимает два атома водорода (X-лиганды) и одна молекула водорода (η2-лиганд L-типа) – такой получается смешанный гидрид. Удивительно, что такая странная штука еще и вполне устойчива. Понятно, что именно жесткий тридентатный хелатор, прочно контролирующий три посадочных места, не оставляет этому комплексу возможностей во что-то другое превратиться. Комплекс очень эффективно катализирует транс-гидроборирование. Терминальный протон при этом переселяется на второй атом углерода, что отлично видно из реакции дейтерированного соединения.
![]()
Если такому комплексу дать пинаколборан, происходит банальное преметаллирование, причем в качестве уходящих групп с обоих сторон выступают водороды (точнее, гидрид и протон). Бор входит в координационную сферу. Нарисуем цикл, условно обозначив хелатный лиганд, и опустив немного запутанный процесс первого входа реагентов в координационную сферу.
- алкин входит и дает винилиден с перемещением протона от первого углерода к второму;
- происходит миграционное внедрение – борильный лиганд перемещается на карбеновый углерод, это обычная реакция для карбенов, винилиден при этом превращается в X-лиганд, степень окисления не изменяется; стереохимия продукта определяется именно на этой стадии, так чтобы с одной стороны не оказался металл вместе со всей своей громоздкой коллекцией лигандов и заместитель из ацетилена;
- дальше может произойти восстановительное элиминирование продукта, и так это и нарисовано, хотя это не однозначно – рутений не очень любит нольвалентное состояние. Для этой стадии может быть альтернатива – расщепление связи Ru-C бораном (это похоже на переметаллирование) и тогда весь цикл пролетит без изменения степени окисления рутения. Это место я оставлю на ваше усмотрение. Строго говоря, это установить невозможно, и выбор делается на основе общих соображений (аналогий из химии других комплексов того же металла).
![]()
Транс-гидроборирование. Теперь и на меди.
В современной органической химии с участием переходных металлов есть один металл, совершенно идеально вписывающийся в идиому “к каждой бочке затычка”. Лет 20 назад таким металлом был палладий, а в последние 10 лет это медь. Можете быть уверены, если есть какой-то процесс с участием комплексов переходных металлов, то рано или поздно туда запихают медь. Вот и в гидроборировании медные катализаторы тоже заявились (Yun et al, Org.Lett., 2016, 18, 1390), причем сразу и в обычном цис-гидроборировании, и в аномальном транс-гидроборировании. ![]() И если протокол для цис-гидроборирования получился громоздкий и неудобный, то транс-гидроборирование заслуживает внимания. Обратим внимание на несколько вещей. Во-первых, в качестве борана взят не обычный pinBH, а довольно хитрый диаминоборан, производное 1,8-диаминонафталина. Так как такая живописная нашлепка на боре вполне годится для последующего кросс-сочетания, хотя и требует специальных протоколов в реакции Судзуки-Мияуры, эта часть протокола вполне терпима, хотя и составляет вопиющее нарушение принципа экономии атомов. В сложном синтезе такой борный остаток, кстати, даже любят, потому что он позволяет выполнять несколько кросс-сочетаний в синтезе сложных молекул с разными борными остатками за счёт использования разных протоколов кросс-сочетания. В качестве исходного комплекса меди взят очень популярный комплекс Липшутца – соль 2-тиофенкарбоновой кислоты, которую везде и всегда обозначают CuTC. В чем секрет этой странной соли не знает кажется и сам проф. Липшутц, однажды использовавший ее в одной из своих работ. Она растворима в органических растворителях (как, впрочем, и многие другие соли меди и карбоновых кислот), вполне устойчива (как, впрочем …), дешевле грязи (как, впрочем …) – все, больше аргументов нет. Просто есть такая соль, и точка. К ней настолько все привыкли, что часто даже не расшифровывают, что это за ТС такое. Реакция идет в присутствии бидентатного фосфина, очень похожего на уже знакомый нам XanthPhos, но более простого.
И если протокол для цис-гидроборирования получился громоздкий и неудобный, то транс-гидроборирование заслуживает внимания. Обратим внимание на несколько вещей. Во-первых, в качестве борана взят не обычный pinBH, а довольно хитрый диаминоборан, производное 1,8-диаминонафталина. Так как такая живописная нашлепка на боре вполне годится для последующего кросс-сочетания, хотя и требует специальных протоколов в реакции Судзуки-Мияуры, эта часть протокола вполне терпима, хотя и составляет вопиющее нарушение принципа экономии атомов. В сложном синтезе такой борный остаток, кстати, даже любят, потому что он позволяет выполнять несколько кросс-сочетаний в синтезе сложных молекул с разными борными остатками за счёт использования разных протоколов кросс-сочетания. В качестве исходного комплекса меди взят очень популярный комплекс Липшутца – соль 2-тиофенкарбоновой кислоты, которую везде и всегда обозначают CuTC. В чем секрет этой странной соли не знает кажется и сам проф. Липшутц, однажды использовавший ее в одной из своих работ. Она растворима в органических растворителях (как, впрочем, и многие другие соли меди и карбоновых кислот), вполне устойчива (как, впрочем …), дешевле грязи (как, впрочем …) – все, больше аргументов нет. Просто есть такая соль, и точка. К ней настолько все привыкли, что часто даже не расшифровывают, что это за ТС такое. Реакция идет в присутствии бидентатного фосфина, очень похожего на уже знакомый нам XanthPhos, но более простого.