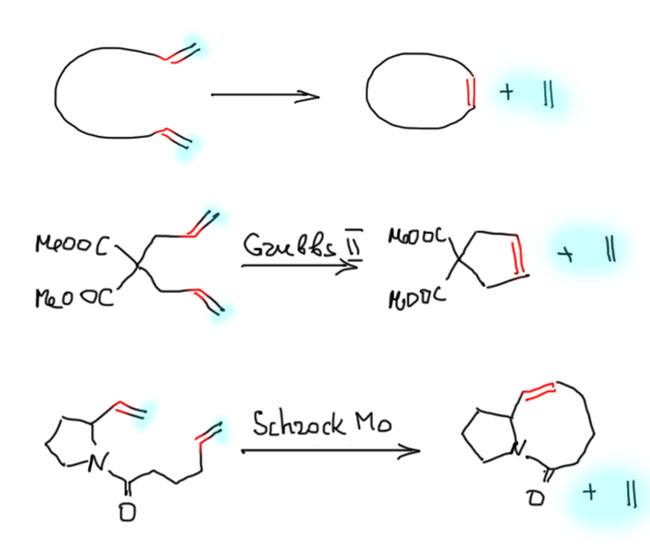
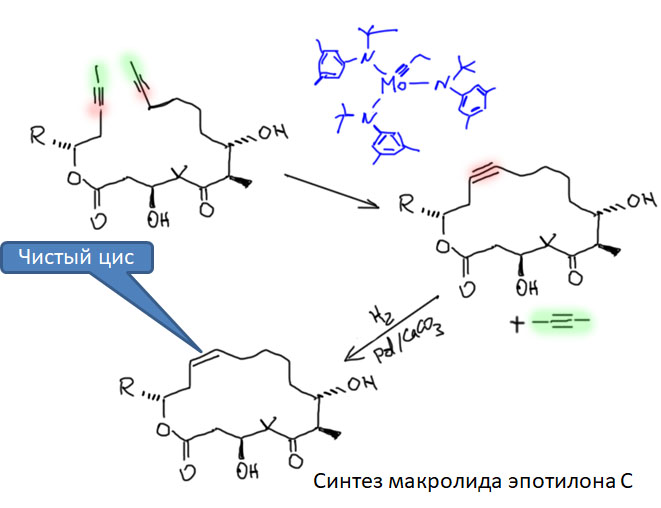
Олефины и ацетилены
Соединения с кратными углерод-углеродными связями являются одними из важнейших субстратов для реакций с участием комплексов переходных металлов. Количество таких реакций огромно, и регулярно увеличивается. Изучение таких процессов – один из самых живых разделов органической химии. Среди реакций олефинов и ацетиленов очень много процессов промышленного значения, и не только в тонком органическом синтезе современных лекарственных препаратов и новых материалов, но и в крупнотоннажном синтезе полупродуктов. Как и в смежных областях химии комплексов переходных металлов в органическом синтезе, в последние 20 лет наблюдается бурный прогресс, связанный с качественным скачком в понимании закономерностей реакционной способности координационных соединений и рационального дизайна лигандов и реагентов новых типов.
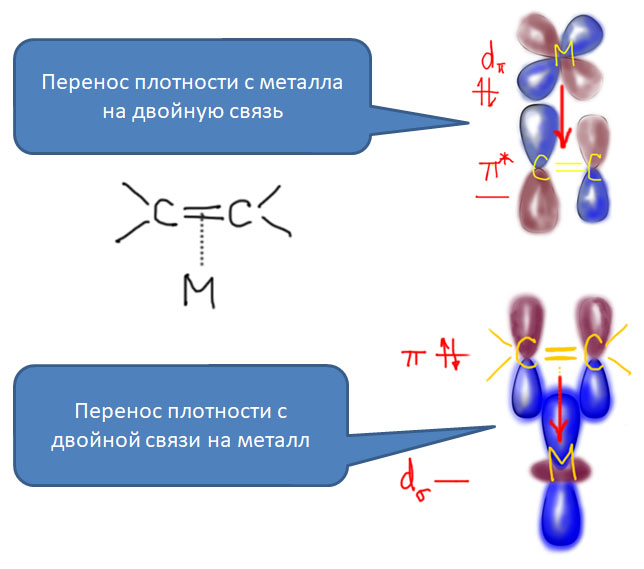
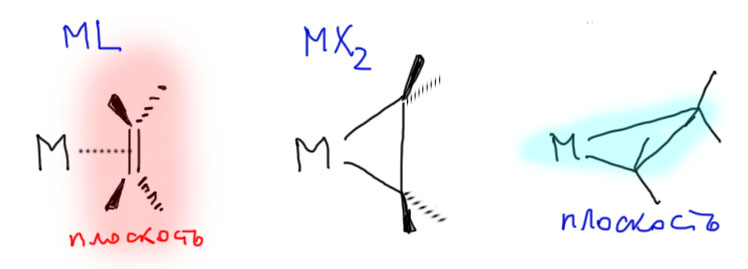
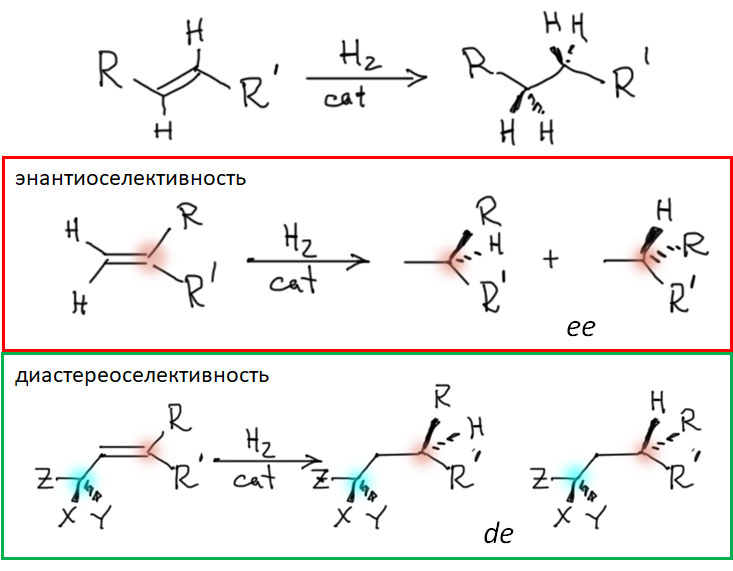
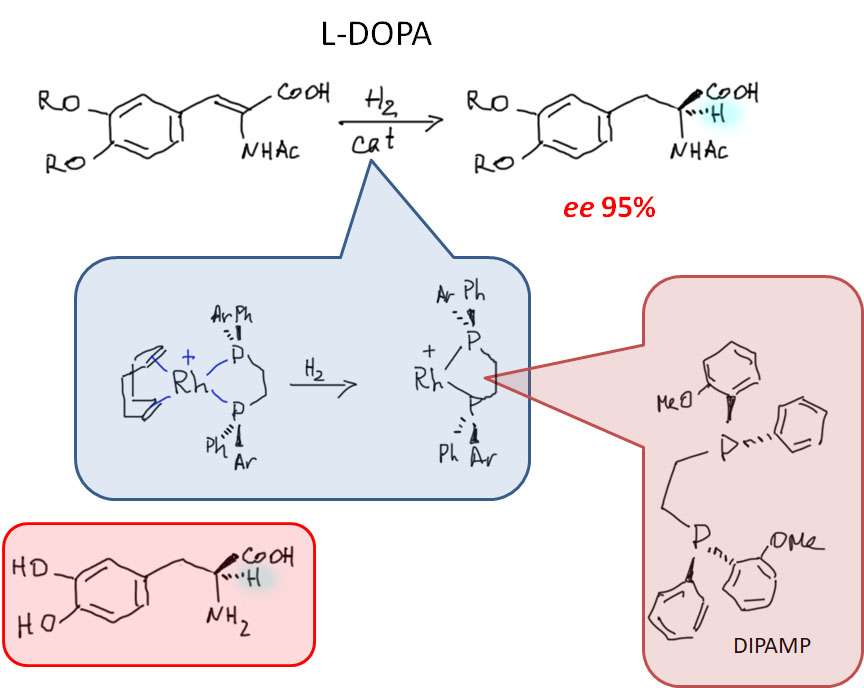
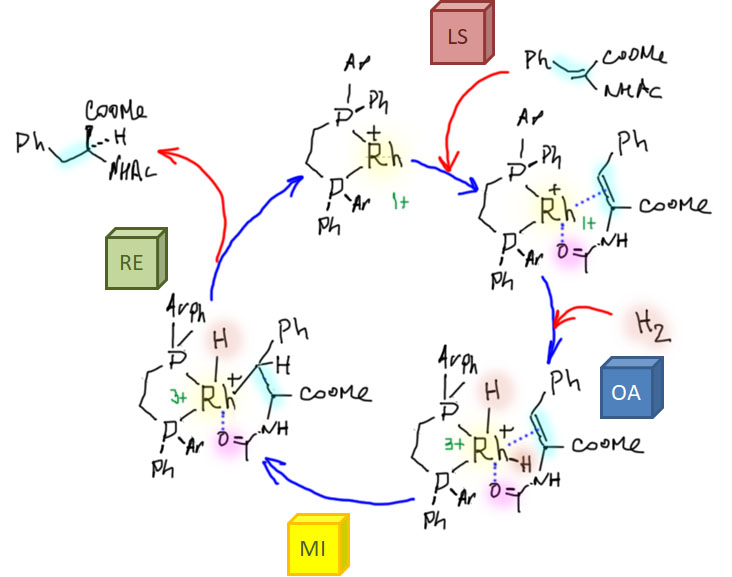
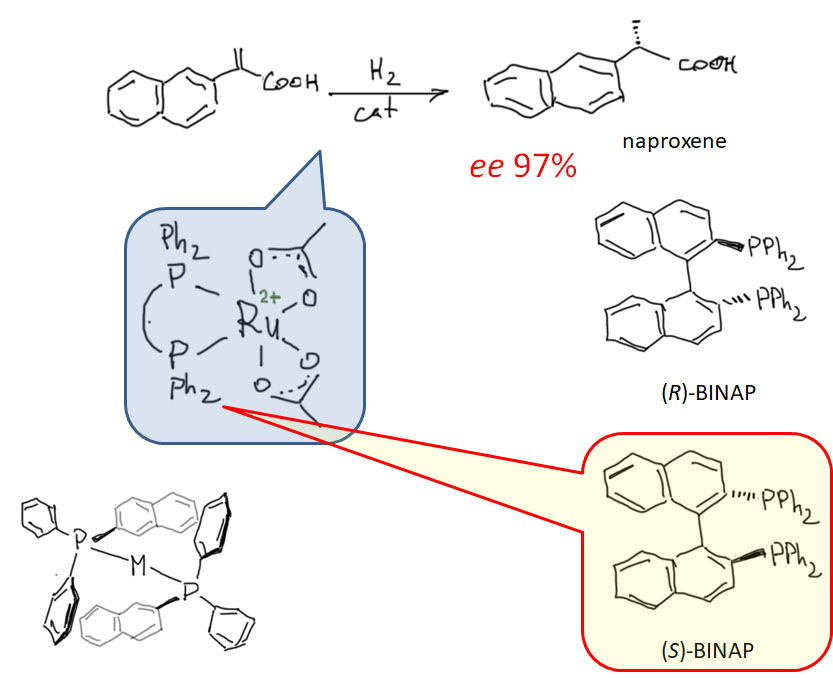
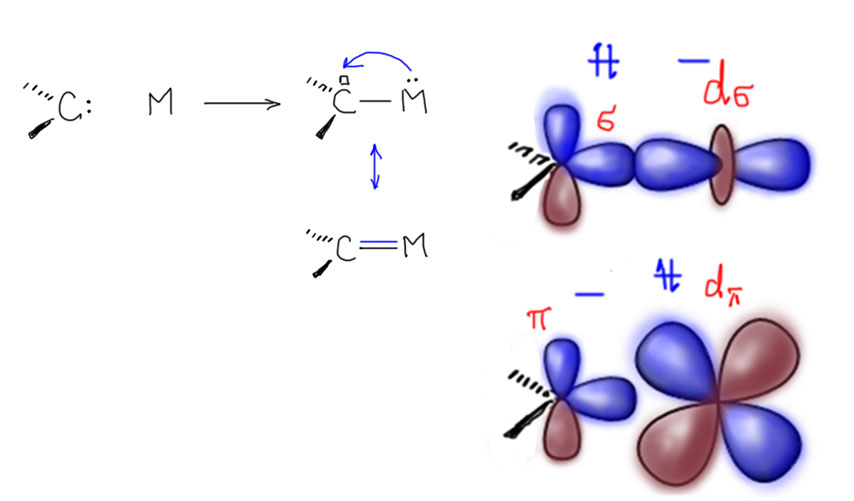
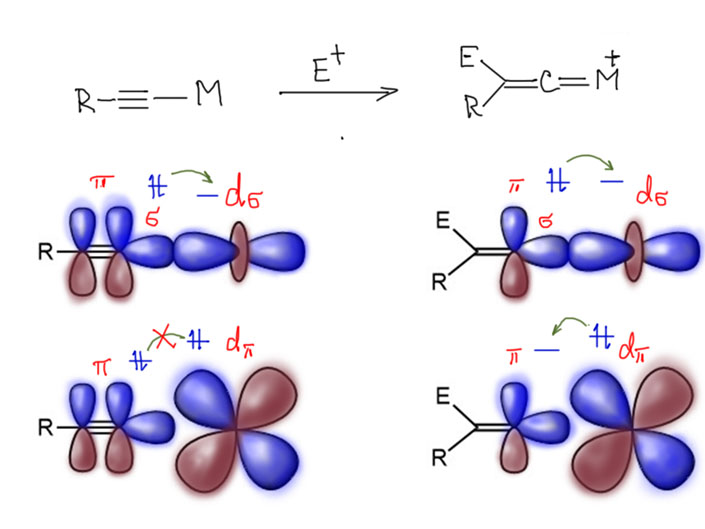
Реакционная способность олефинов и ацетиленов после вхождения в координационную сферу металла определяется тонким балансом двух взаимодействий – прямой координационной связи и эффекта обратного донорного взаимодействия, отвечающие за противоположные направления смещения электронной плотности – от лиганда на металл и от металла на лиганд. Сначала посмотрим на два крайних случая, когда преобладает одно из взаимодействий. Преобладание обратного донорного эффекта (back-donation) сообщает координированному олефину свойства нуклеофила, а каждый из двух атомов углерода связи становится похож на карбанион.
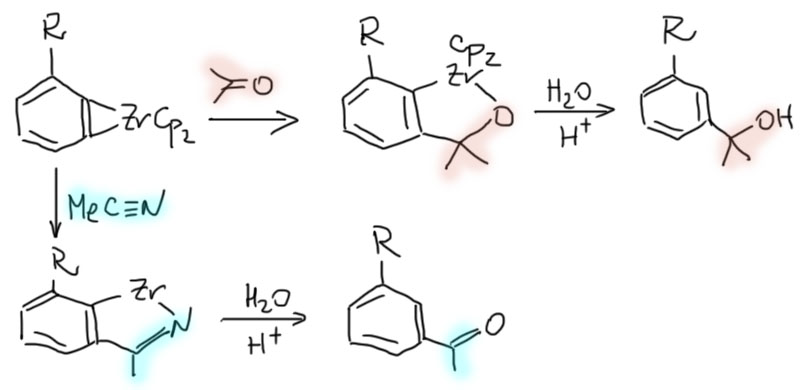
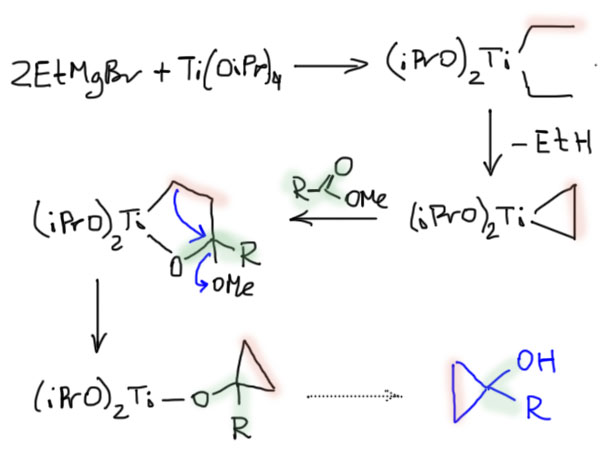
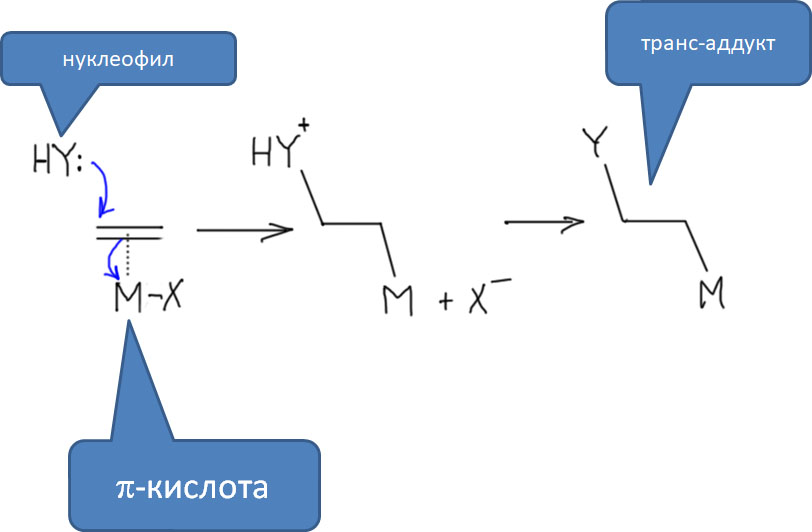
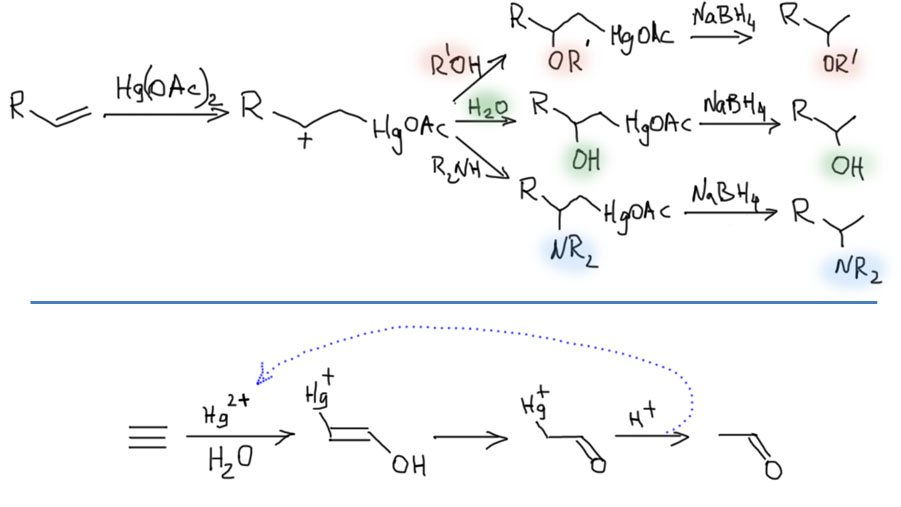
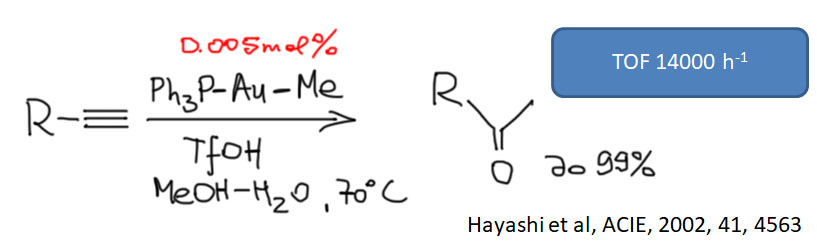
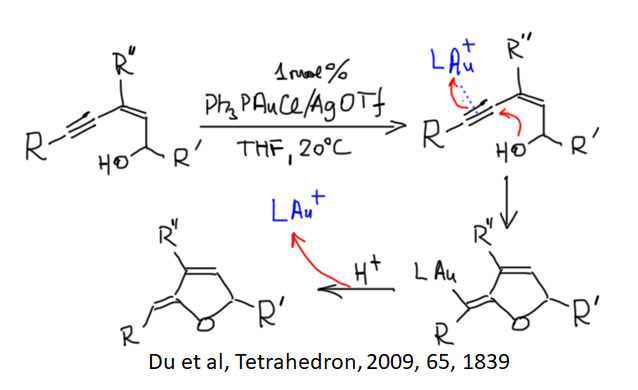
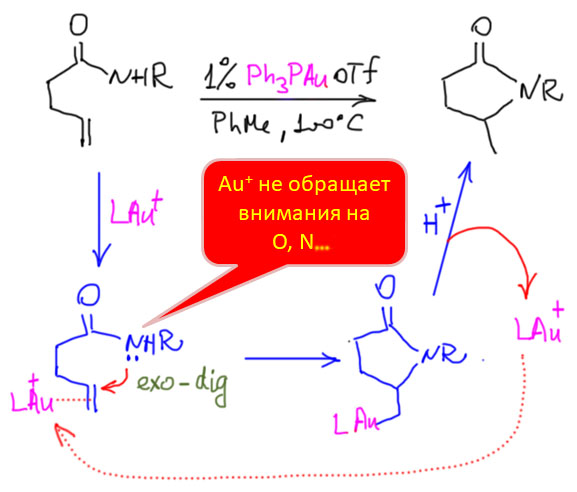
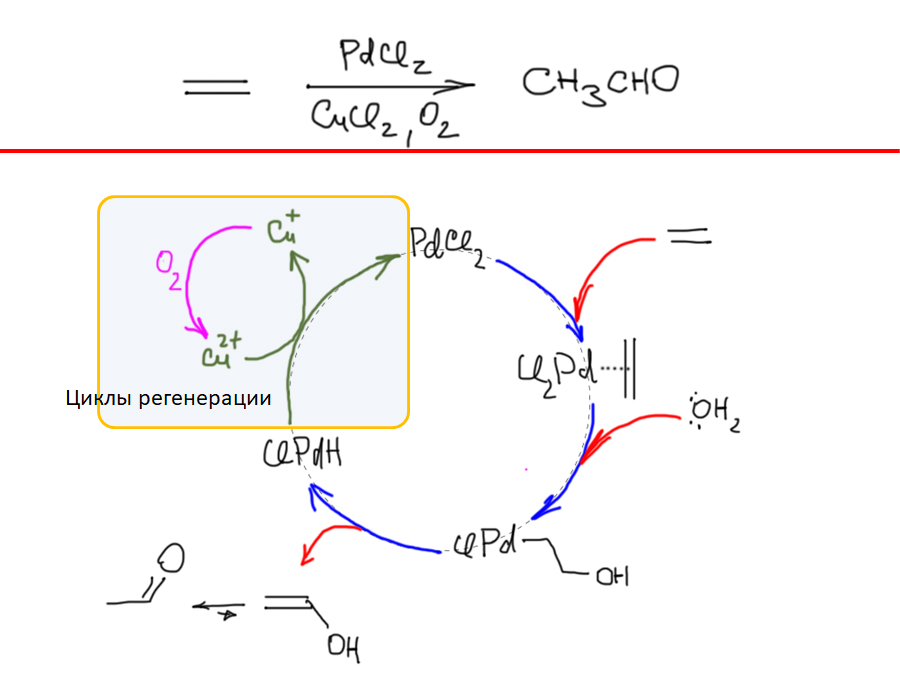
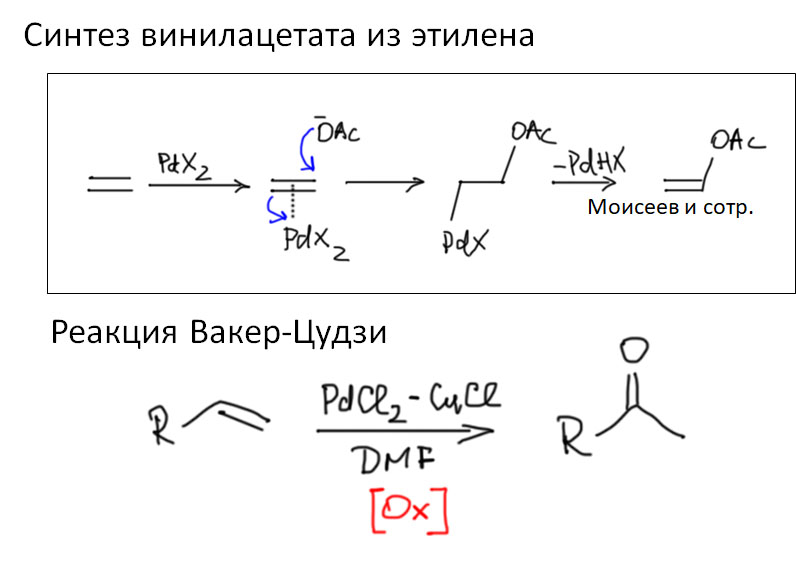
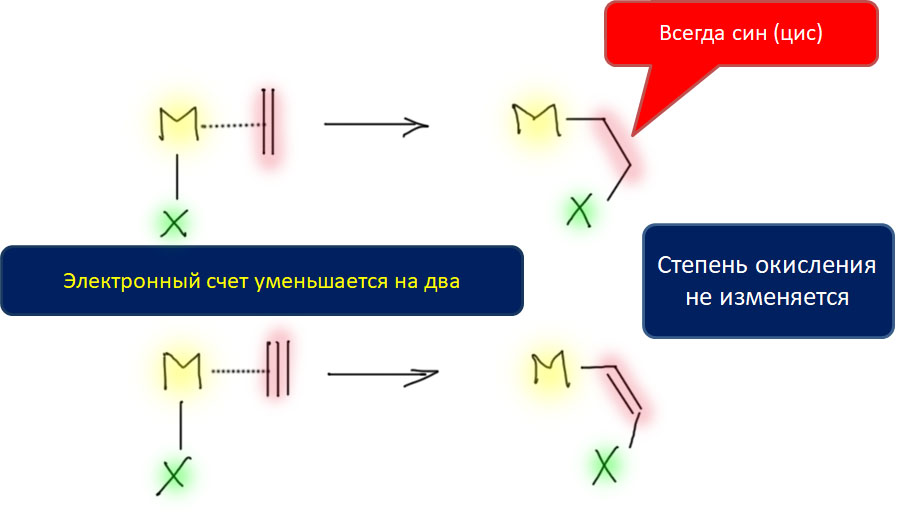
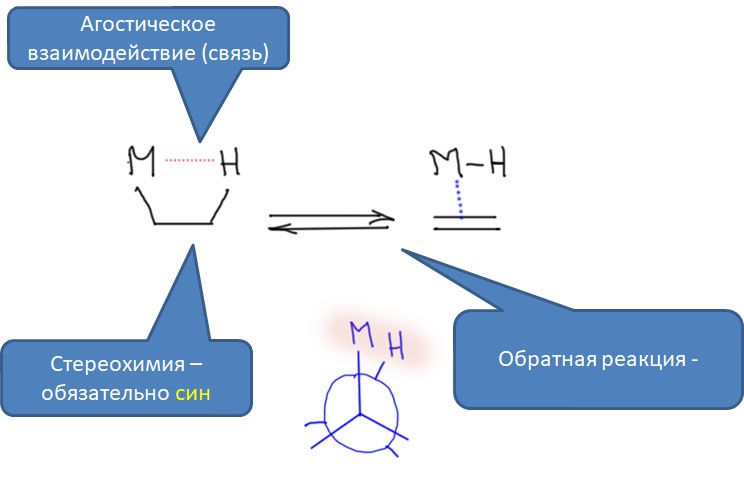
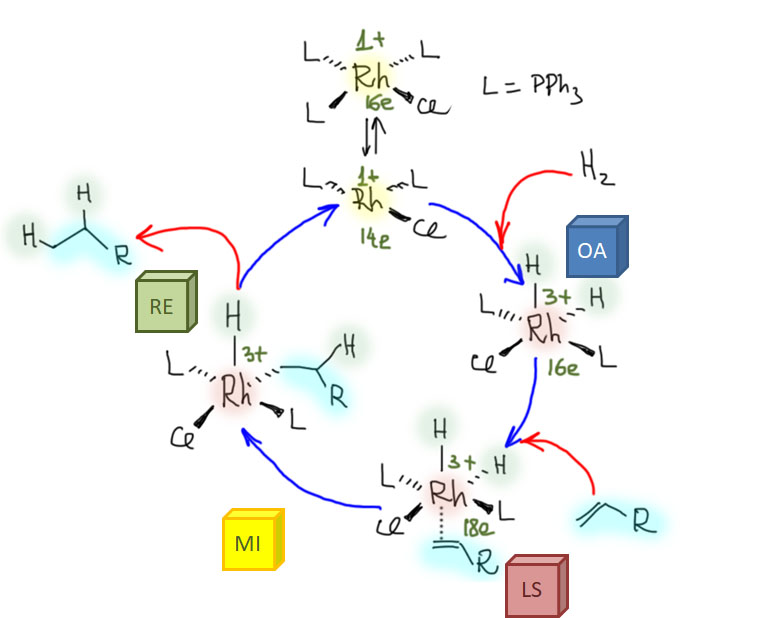
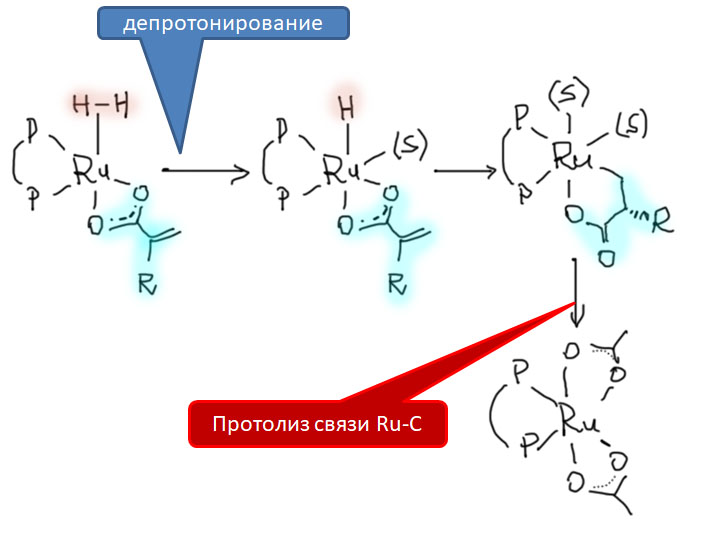
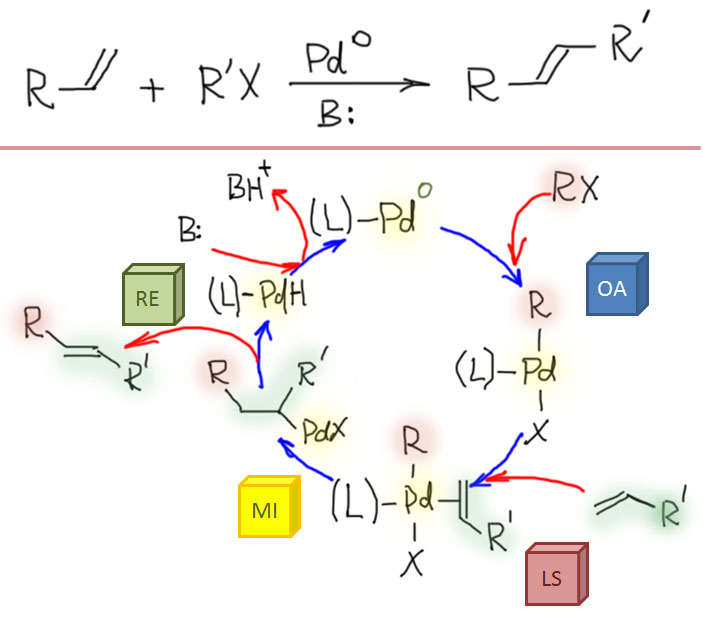
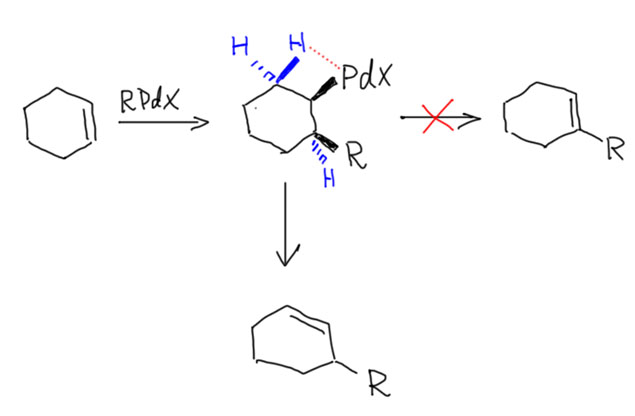
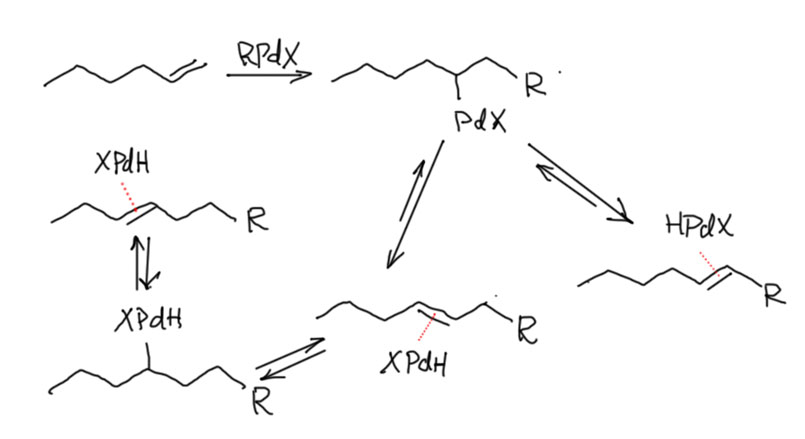
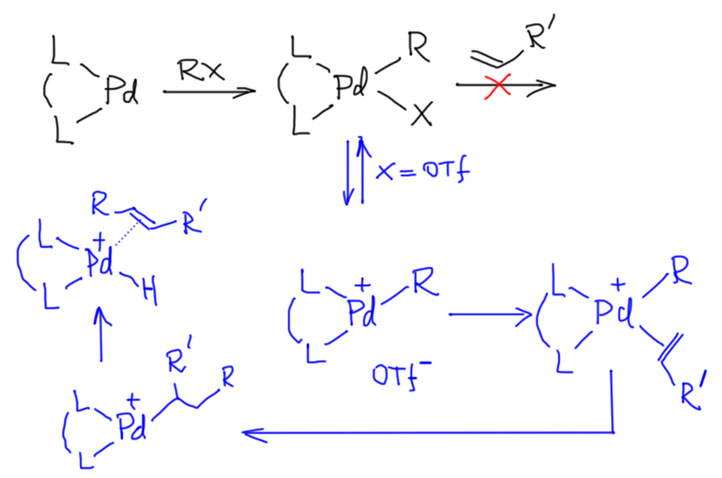
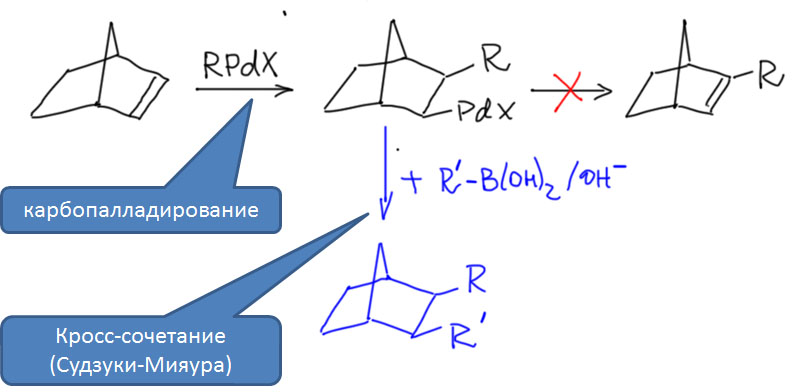
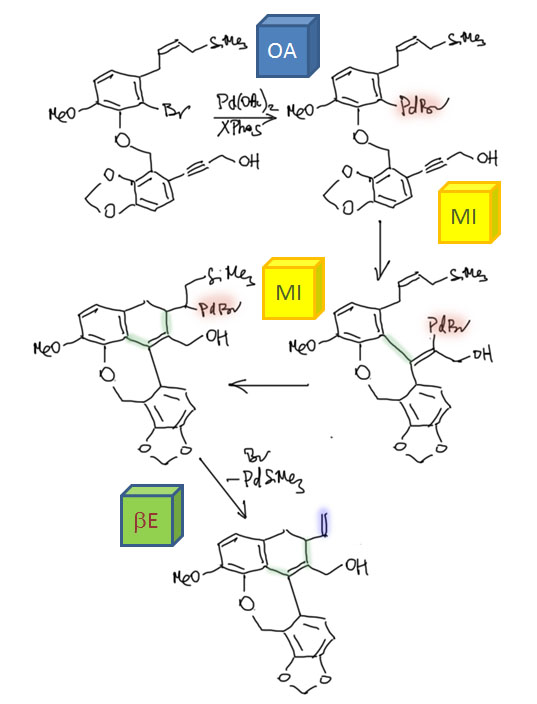
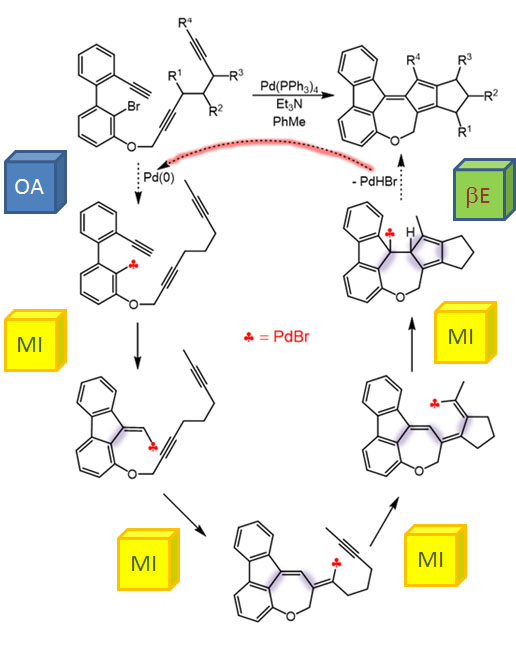
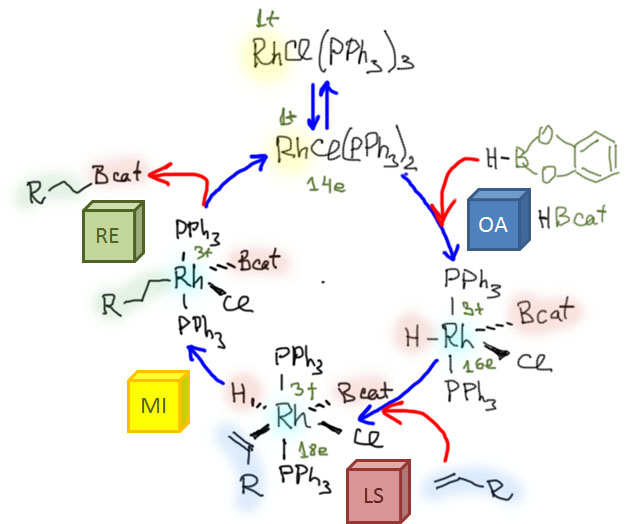
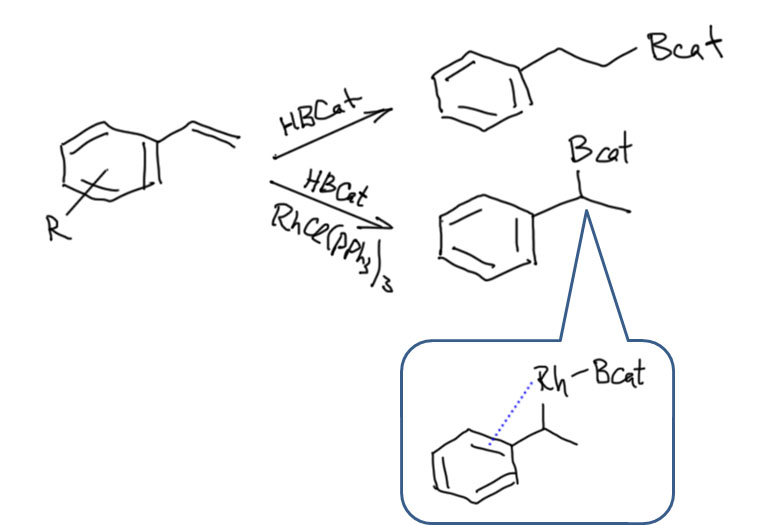
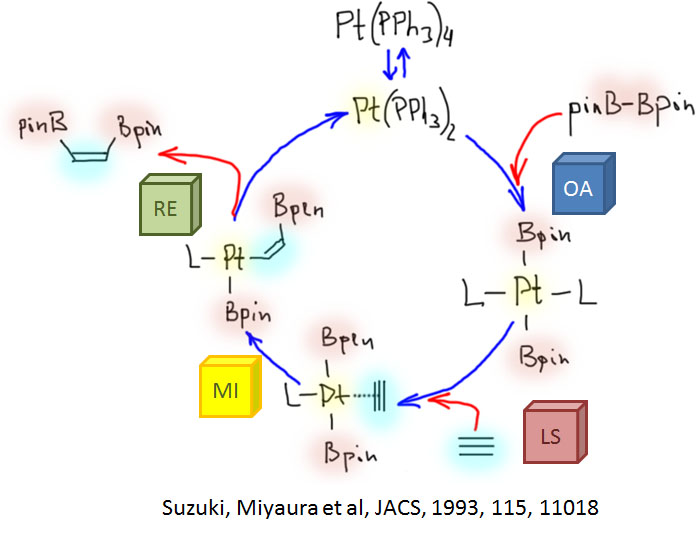
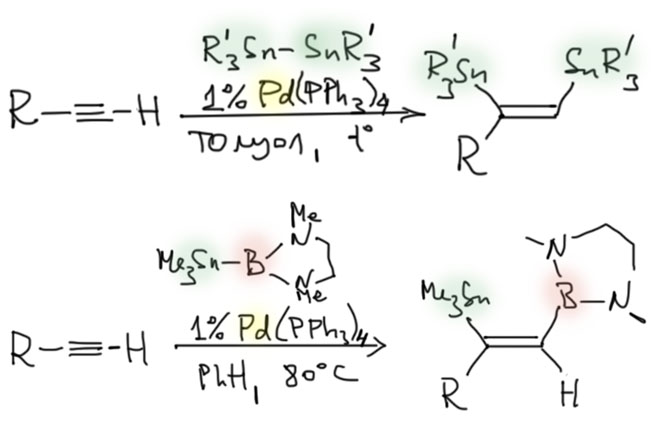
Реакции с участием комплексов карбенов и карбинов
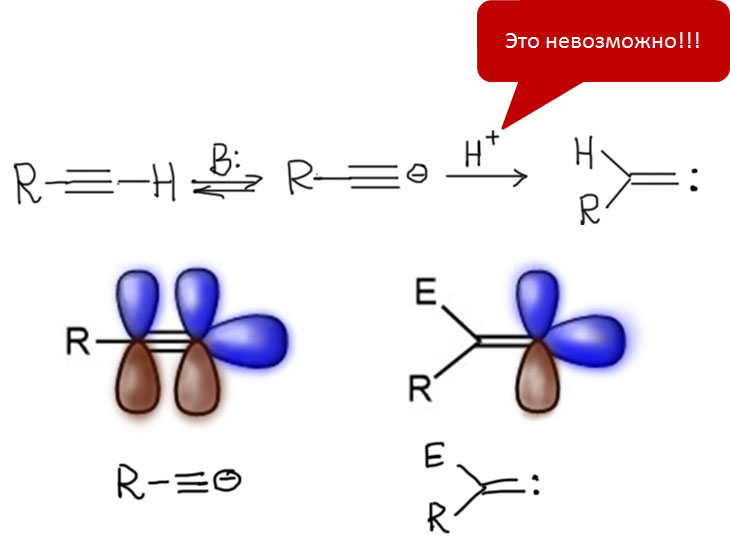
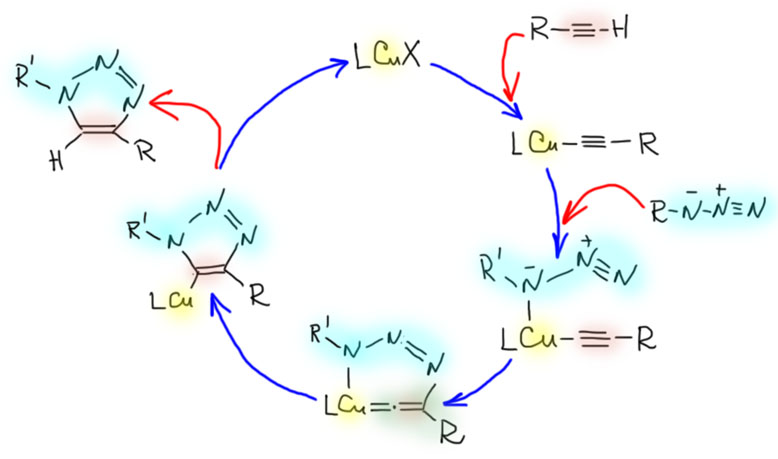
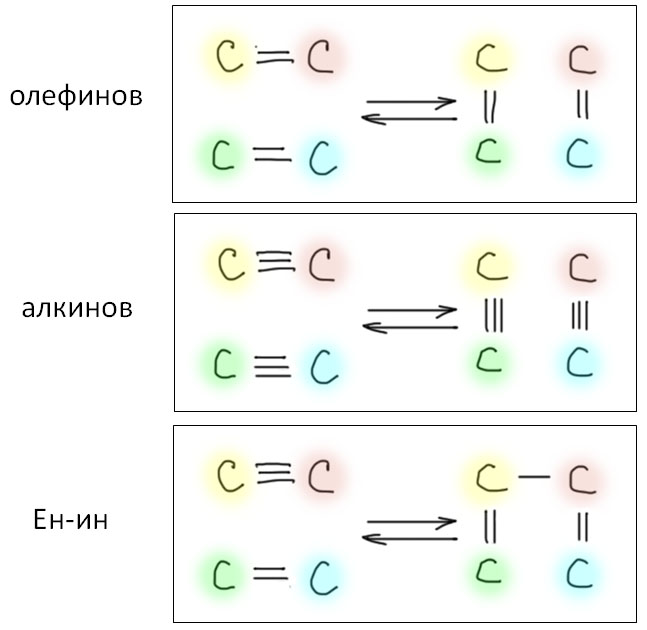
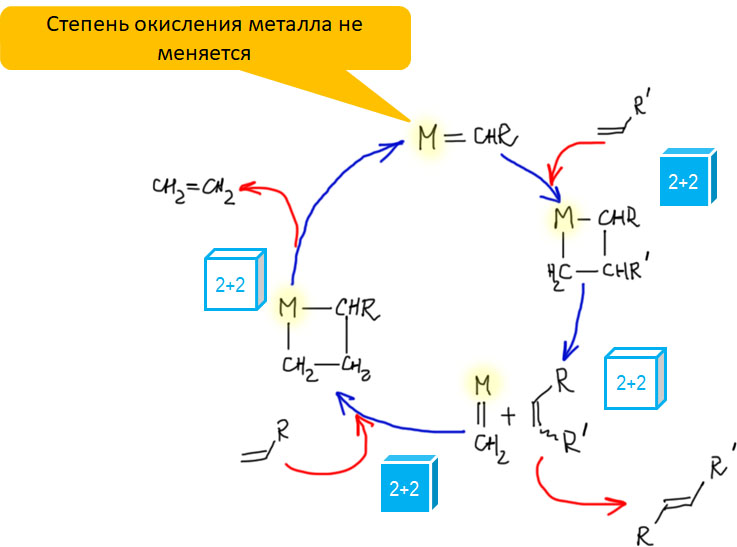
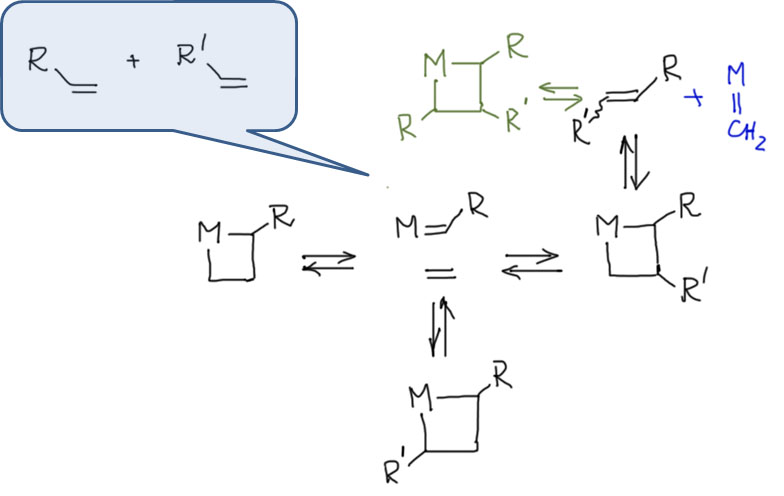
Карбен – это половинка олефина, а карбин – половинка ацетилена. Комплексы карбенов и карбинов очень часто встречаются в реакциях непредельных соединений с участием комплексов переходных металлов, поэтому и заняться ими имеет смысл именно в этом разделе.

Рутениевые катализаторы
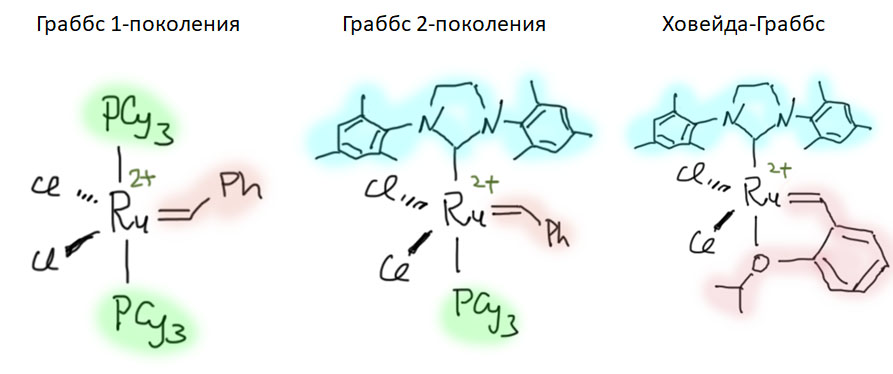
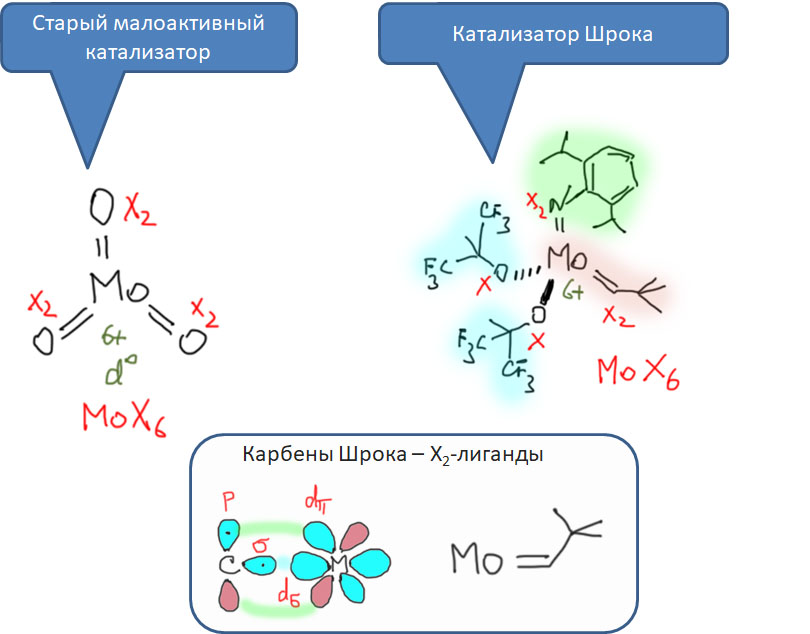
Чаще всего применяются в метатезисе олефинов более устойчивые и удобные катализаторы на основе позднего переходного металла рутения. В таких комплексах карбены – L-лиганды. Несколько поколений таких комплексов предложил Граббс и его последователи. Замена фосфина карбеновым лигандом и хелатирующим карбеном повышает активность комплексов (TOF) и их долговечность (TON).