метатезис олефинов
DF
Любопытный пример метатезиса с претензией на новый тип этого превращения – RRM, Ring Rearrangement Metathesis. Разберёмся сначала в самой реакции, а потом попробуем понять, есть ли за этими претензиями что-то существенное.
В статье описаны два диастереомера исходного субстрата – экзо и эндо, но в задании надо было найти конкретный. Если не смогли и взяли первый попавшийся, не обижайтесь, плюсиков получите немного. В органике нельзя пренебрегать стереохимией, это просто непрофессионально. Диастереомеры – разные соединения с разной структурой и реакционной способностью, и если нужно сделать один, бесполезно предъявлять другой и ссылаться на то, что не рассмотрели, похожи как собаки, какая разница, в следующий раз буду внимательнее, мне вообще органика до лампочки, понарисуют тут всяких раскоряк а мне глаза портить и т.д. Это не то и всё тут!
Итак, исходное это явно аддукт Дильса-Альдера циклопентадиена, дважды аллилированный, в виде стереохимически чистого диастереомера, являющегося мезо-формой (плоскость сечёт всю молекулу через вершинки мостиков и в этой плоскости лежат метил и бензил) и оптически неактивного, следовательно никакой энантиоселективности здесь искать нельзя, а диастереоспецифичность задана тем, что стереоцентры не затрагиваются. Реакцию вели в присутствии одного из двух классических катализаторов Граббса в количестве 10 и 20 моль %. Диастереоизомеры по разному откликнулись на эти предложения, что еще раз напоминает нам о важности стереохимии даже в тех случаях, когда она вроде бы и не затронута, но это не так – форма молекул различается, хотя бы по отношению вновь возникающего кольца и мостика. Предсказать тут ничего нельзя. Возможно, помогло бы кропотливое моделирование путей реакции, но кто ж такие вещи будет делать.
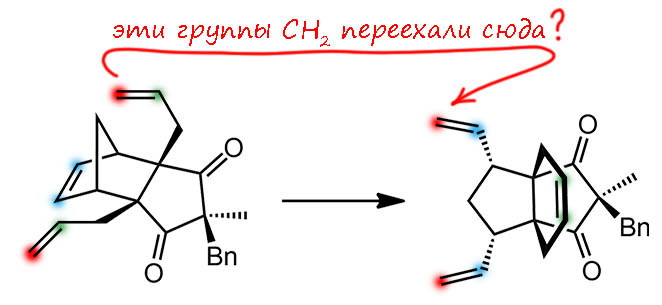
Нужный нам диастереомер предпочёл старый добрый граббс-один – а вы думали, что его на помойку выкинули, после того, как с большой помпой разработали и внедрили новые поколения катализаторов? Нет, жив здоров и еще отметит столетие добросовестной службы и стотысячный продукт, полученный с его участием в реакциях, где он шутя обштопает все модные новинки. Классика всегда в моде. Граббс-два, кстати тоже не юноша, отлично справился со вторым диастереомером. Результат оказался сильно зависим от загрузки катализатора и выглядит это довольно странно – при 10 моль% оба продукта получаются поровну, в сумме 80% при 20 моль% преобладает продукт по обоим направлениям, а в сумме продуктов 87%. Прикинем TON. Как мы знаем, в метатезисе обычное определение каталитического цикла не очень адекватно, так как реакция фактически бесконечна (прерывается гашением реакционной смеси) и мы имеем дело с установившимся или не успевшим установится равновесием. Тем не менее с чисто практической точки зрения можно принять, что выход продукта по каждому завершившемуся акту метатезиса и дает нам формально количество циклов. Тогда, по второму продукту, где акт метатезиса один (замкнулся цикл), цикл один. По основному продукту, где акта метатезиса два (замкнулся один цикл, разомкнулся другой) мы имеем около 7 циклов (каждые 20% выхода образутся за 2 цикла). Итого TON около 8. Всё это удалось достичь за 10 часов, имеем приблизительно цикл в час. Небыстро. Реакция идет при комнатной тепературе при довольно низкой концентрации реагентов. Но катализаторы Граббса сделаны для удобства, а не для скорости – они и правда не очень активные. Дело делают, много денег не просят, и хорошо. Не нравится – разрабатывайте новые. 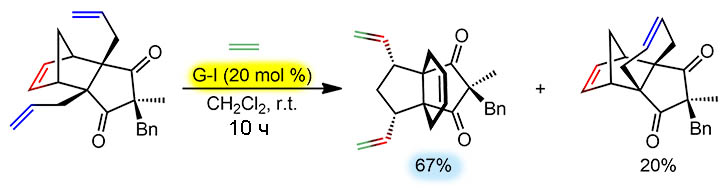 Теперь посмотрим, а что тут действитеьно случилось. Второй продукт оставим – это просто обычный RCM на шестичленный цикл, первое, что ожидается. Первый продукт интереснее, хотя он легко читается как сначала тот же RCM, который явно идёт быстрее, и дальше метатезис раскрытия цикла, обратный к RCM. Но – если просто выписать исходное и этот продукт, оказывается, что это изомеры, значит превращение одного в жругой это формально перегруппировка (rearrangement) и тогда почему бы это не назвать так, как это сделали авторы статьи – ring rearrangement, то есть перегруппировка циклов. Вот просто пометим цветом атомы, участвующие в деле и тогда увидим, что синенькие и зелененькие остались на месте, а красненькие как будто переехали из одного места в другое. Ну ничего себе! Действительно невероятно! Новый тип метатезиса. А почему журнал такой скромный? Это же ангевандте или джакс без разговоров, с руками оторвут и ещё попросят. Вот, индусы какие скромные.
Теперь посмотрим, а что тут действитеьно случилось. Второй продукт оставим – это просто обычный RCM на шестичленный цикл, первое, что ожидается. Первый продукт интереснее, хотя он легко читается как сначала тот же RCM, который явно идёт быстрее, и дальше метатезис раскрытия цикла, обратный к RCM. Но – если просто выписать исходное и этот продукт, оказывается, что это изомеры, значит превращение одного в жругой это формально перегруппировка (rearrangement) и тогда почему бы это не назвать так, как это сделали авторы статьи – ring rearrangement, то есть перегруппировка циклов. Вот просто пометим цветом атомы, участвующие в деле и тогда увидим, что синенькие и зелененькие остались на месте, а красненькие как будто переехали из одного места в другое. Ну ничего себе! Действительно невероятно! Новый тип метатезиса. А почему журнал такой скромный? Это же ангевандте или джакс без разговоров, с руками оторвут и ещё попросят. Вот, индусы какие скромные.
Немного ещё потерпим петь дифирамб мощным индийским исследователям и попробуем нарисовать механизм – ведь метатезис работает всегда одинаково, надо только аккуратно расписать возможные варианты комбинации карбена и двойных связей. Поскольку субстрат очень симметричен, рисовать придется не так много, ведь многие пути повторяют друг друга. Итак, опустим стадию инициирования и предположим, что весь рутениевый карбен, ведущий реакцию, взялся из реакции исходного предкатализатора с субстратом. Тогда метилен всегда будет красненьким. Видим, что сначала идёт RCM, а метиленовый комплекс дальше ведёт метатезис циклизации. Всё на месте, красненькие встали – ровно так как нарисовано на схеме перегруппировки. Вот она, слава!
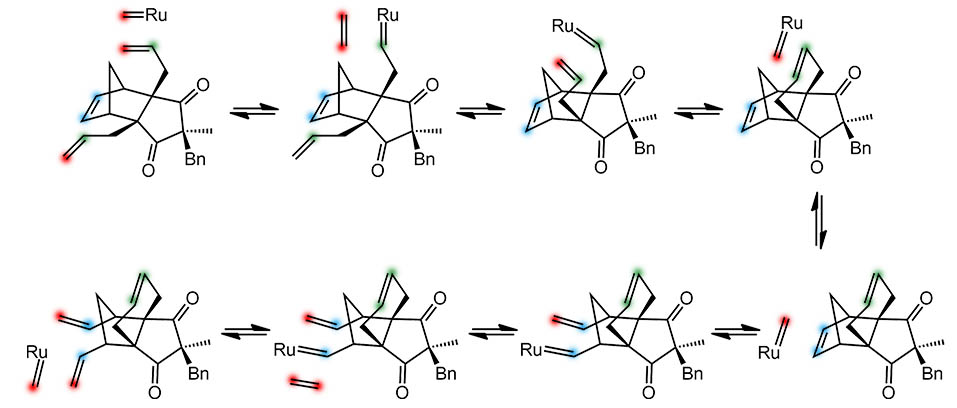
Постойте, но там на схеме реакции ещё кое-что написано. И в методике указано. Реакцию ведут в атмосфере этилена. Этилена? Зачем? Ведь в RCM этилен выделяется. Да, но сколько его там. Субстрата очень мало, этилена даже при количественном выходе RCM получится – с одной стороны ровно эквивалент, но с другой стороны это очень малая концентрация, учитывая еще и то, что он будет выходить в газовую фазу, уходя из раствора – значит, пришлось бы городить прибор типа шприцевого реактора без газовой фазы, и тогда ждать, что вторая реакция пойдёт с такой малой концентрацией этилена. Но там есть конкурент – обычная метатезисная полимеризация ROMP, которая отлично идет с такими структурами и с которой вообще всё начиналось когда-то. Понятно, что этого конкурента надо давить. Чем? Да просто концентрацией этилена, чтобы его было достаточно и рутений хватал бы именно его просто по причине большого избытка. И избыток такой будет, если реакцию вести в атмосфере этилена. Ровно так, как делают индийские исследователи. Но тогда, в карбеновом комплексе почти весь метилен будет из чужого этилена – пометим его желтеньким. Просто по теории вероятностей и законам равновесий. И тогда схема немного изменится. Механизм останется тем же, но красненький цвет довольно быстро вытеснится желтеньким из добавленного этилена. И второй метатезис станет просто обычным раскрытием цикла – ring opening metathesis, ROM.Это больше не перегруппировка, а просто два метатезиса, один за другим, и это не каскад, потому что на первом можно остановиться, и так и получается второй продукт. Это типичный тандем, в данном случае RCM-ROM.
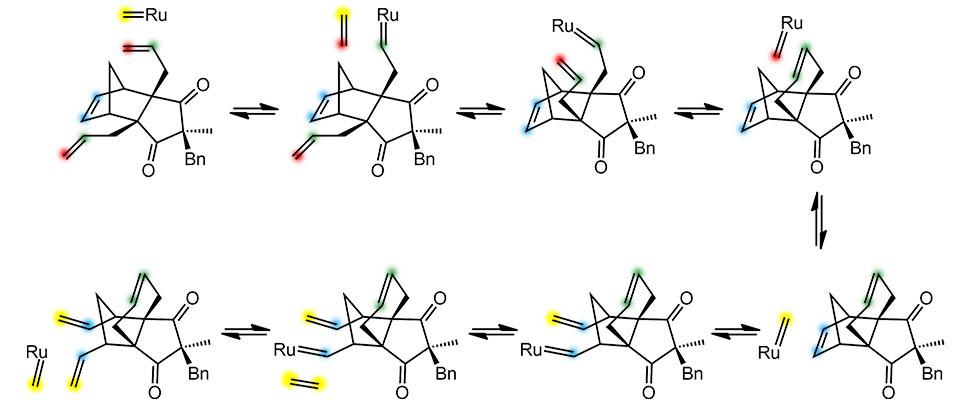
Забавно, но не тянет на гламурные журналы. И никакой это не новый тип метатезиса, а два старых, в тандеме. Отлично можно догадаться, что авторы работы пытались пропихнуть это либо в ангевандте, либо хотя бы в один из парных журналов Chemistry European Journal/Asian Journal, тоже довольно гламурных, и их оттуда выперли в этот новый журнал, который для того и завели совмем недавно, чтобы приличные работы (а это приличная работа, хотя и с немного странным экспериментом) из гламурных журналов издательства Wiley не терять, а пристраивать в приличный журнал того же издательства. Обычно, если отзывы рефери из гламурных журналов такие, что ясно, что работа хорошая, но не обещающая большого цитирования, процесс перенаправления происходит автоматически. Не плачь, дорогой автор – подразумевает редактор гламурного журнала – статью твою мы напечатаем, у нас тут журнальчик есть новый, красивый, обложка почти такая же (правда импакт в несколько раз меньше и не растет, собака) – не надо в другое издательство, там тебя тоже бортанут, ведь сам понимаешь, что никакого нового метатезиса тут нет, да и эксперимент какой-то простоватый, уж хотя бы изотопную метку добавил, чтобы показать, что хотя бы иногда метиленовая группа и правда мигрирует внутри молекулы. Авторы всё поняли и не стали возражать, хотя название не изменили.
Последний вопрос – почему происходит то, что происходит – один цикл закрывается, другой раскрывается. Это просто – при этом уменьшается напряжение. Исходная система норборненового типа, в ней есть напряжение, два цикла склеены посредине мостиком, это заставляет углы отклоняться от нормальных значений, плюс тут немалое торсионное напряжение, так как нижняя часть это пресловутая ванна с заслоненными конфигурациями по связям. После реакции этопросто три хороших цикла – пять, пять, шесть – соединённых по связи, такие системы принято называть пропелланами, и они становятся весьма нетривиальными, когда хотя бы часть циклов была бы поменьше – 4 или 3. Но такая система вполне проста и лишена серьёзных напряжений. Поэтому и получается с довольно приличным выходом, хотя ROM редко бывает эффективным из-за конкуренции с полимеризацией.
DL
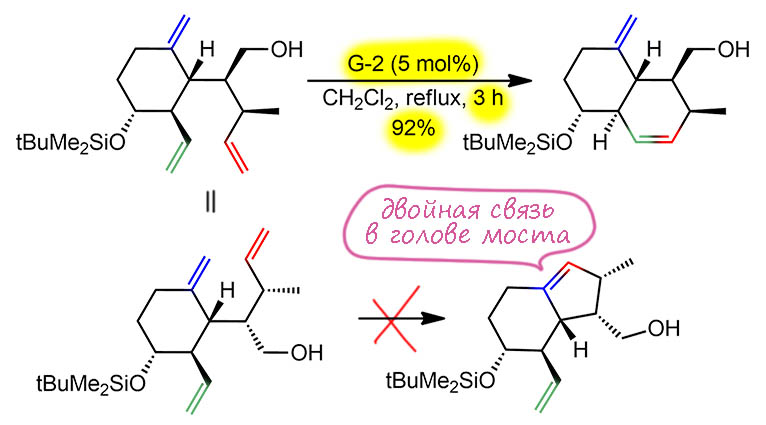
Циклизация метатезисом (RCM) в середине длинного синтеза очередных антибиотиков. Довольно простой случай, всё очевидно – два терминальных олефина, образуется шестичленный цикл. Авторы выбирают граббс-два без попыток оптимизации, видимо, просто любят этот катализатор и доверяют ему, стоит на полке и раньше не подводил, хороший. И сейчас не подвёл – выход 92% при загрузке катализатора 5 моль% – соответственно TON около 18, вполне типично для метатезиса. Реакцию вели при слабом нагревании – кипение дихлорметана это 45 градусов, 3 часа. TOF – 6 циклов в час, цикл каждые 10 минут, неплохо. В субстрате полно стереоцентров в определённой конфигурации, и ни один не пострадал, хотя некоторые прямо рядом – это превосходное свойство современных катализаторов метатезиса – они не лезут, куда не просят, хотя тот же рутений, как и вообще вся троица суперметаллов катализа из второго ряда – RuRhPd – очень любит баловаться с кратными связями, двигать их туда-сюда, с неизбежной рацемизацией по следу смещения. Но катализаторы метатезиса хорошо спроектированы, металл обвешан правильными лигандами, которые не только делают его эффективным именно в метатезисе, но и блокируют всякие побочные безобразия.
Единственная проблема, которая здесь еще заслуживает комментария – селективность. В субстрате есть ещё одна двойная связь, которая вполне могла бы в том же метатезисе дать тоже отличный пятичленный цикл, но такой продукт не образуется (даже если его просто не определили, его выход не мог быть выше оставшихся 8%). Почему? Ответ простой: метатезисом рулит стерика и напряжения в циклах. Метатезис строго предпочтёт терминальный винил – однозамещенный этилен – дизамещенному алкену, хоть геминальному, хоть вицинальному. Во-вторых, цикл образовался бы пятичленный, но с двойной связью экзоциклического типа (она же – в голове моста), принадлежащей олновременно двум циклам. Хоть в 5-6 членных циклах это несмертельно, но напряжение вносит, потому что мы вынуждены гнуть углы на sp2-гибридном углероде. Для метатезиса это более чем достаточно, чтобы этот путь заблокировать.
DN
Типичный RCM, в этой работе уже второй – первый макроцикл закрыли тем же RCM ближе к началу синтеза. Там проблем было меньше, потому что в этот момент молекула только начинала строиться и была поменьше. Ближе к концу потребовалось замкнуть второй цикл, и там появились уже потенциальные проблемы – новая двойная связь возникала через атом от уже существующей, а в таких случаях всегда возникает соблазн смещения – чтобы получилось сопряжение. Поэтому система стала сложнее. Мы вообще привыкли, что в метатезисе все просто – исходное и катализатор, положить в кастрюлю, залить растворителем и тушить на медленном огне три-четыре часа – всё, готово, выгружай. Ничего лишнего и доставать продукт просто. Но вот реальный случай – в кастрюлю зачем-то еще положили бензохинон, он желтенький и красивый, но воняет совершенно безбожно. Зачем? И если посмотреть методику, там еще в конце реакци в ту же кастрюлю кладут калийную соль изоциануксусной кислоты и томят варево ещё полчаса. А это зачем?? Разберемся и в том, и в другом – эти приемы стали часто использовать в метатезисах, когда задача хоть чуть сложнее, чем просто закрыть цикл в простой молекуле.
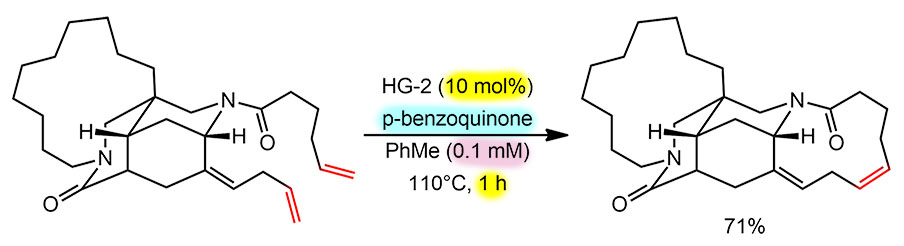
Сначала обратим внимание на то, что реакцию вели при высоком разведении, или, что то же самое – при очень низкой концентрации субстрата: 4 мг – это всего 9 микромоль – в почти 100 мл раствора, то есть если бы потребовался миллимоль, то есть всё равно очень мало, то раствора пришлось бы взять 10 литров – а это уже в лаборатории сделать одной реакцией почти невозможно, надо было бы делить на много повторных замесов! Ну или переходить в пилотное оборудование. Это очень наглядно показывает, какова цена макроциклизации – чтобы направить реакцию по пути мономолекулярной циклизации, а не бимолекулярной димеризации и далее в олигомер, приходится очень сильно разбавлять реакционную смесь, чтобы очень сильно уменьшить вероятность встречи двух молекул субстрата (или, что то же самое, скорость бимолекулярной реакции, зависящей от концентрации в квадрате); скорость макроциклизации (мономолекулярной реакции, где зависимость от концентрации линейна) при этом естественно тоже уменьшается, но не так сильно.
Обычные параметры: при выходе в 71% и загрузке в 10 моль% TON около 7 циклов, все это за час, соответственно и TOF 7 циклов в час. Для такого высокого разведения не так плохо, но приходится делать реакцию в кипящем толуоле, иначе, видимо, слишком медленно. Метатезис для образования хороших циклов (5 или 6) обычно делают при комнатной температуре, потмоу что там не нужно брать такую малую концентрацию. Цис-изомер здесь получается по стереохимическим причинам – хоть это и макроцикл, но неполностью гибкий, в нем есть и еще одна двойная связь, и карбонил, и несколько атомов встроены в жесткий каркас – то есть гибкой остается только небольшая часть и двойные связи уже сближены, так что даже не совсем понятно, зачем было так сильно разбавлять.
Теперь приступим к этим странным добавкам.
В этой статье есть несколько фишек, ставших очень популярными в рутениевом метатезисе. Одна из них – добавление бензохинона для подавления изомеризации, заслуживает подробного разбора и он будет ниже. Вторая, чисто прикладная, удобная методика завершения реакции. С рутениевым метатезисом есть проблема и эта проблема – сам рутений, металл, комплексы которого просто одержимы неукротимым желанием что-нибудь прокатализировать, ну хоть что-нибудь. Я однажды сам с этим столкнулся, когда захотел немного побаловаться с рутениевыми порфиринами и подошёл к делу весьма наивно – и убедился, что с рутением нельзя работать нашим обычным способом «из общих соображений» и «а куда он денется» – пока я щёлкал клювом, разглядывая спектр поглощения в поисках признаков образования металлопорфирина, рутений выдрал CO из растворителя, метанола, и принялся деловито разбирать мой порфирин на части, так что н только металлопорфирин не появлялся, но и сам исходный порфирин стал куда-то исчезать. Ну его ко всем чертям, подумал я тогда и никогда больше с рутением не связывался, бешеный он какой-то. Кстати, это ведь так именно и было – из Большой Троицы катализа RuRhPd рутений пошел в дело позже всех, потому что пришлось долго разбираться, как его укрощать правильными лигандами, элемент этот невероятно строптив и необуздан, прямо как страна, в честь которой его назвали.
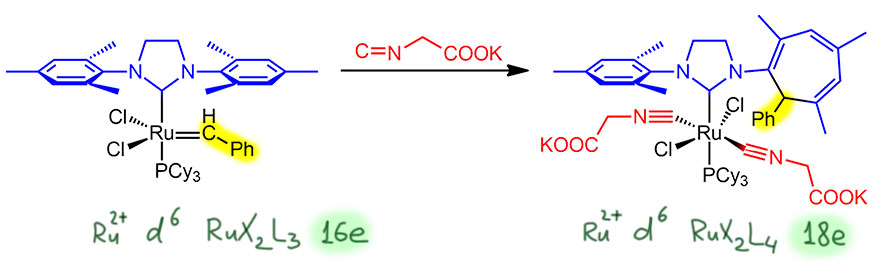
Но вот в метатезисе тоже есть эта проблема – в самой реакции работает катализатор, аккуратно собранный Граббсом и его последователями так, чтобы он ни к кому больше не приставал, но вот реакция закончилась и надо ее, как это принято говорить, погасить. В этот момент, рутениевые комплексы теряют свою структуру, превращаются в какие-то неопознанные соединения, эти рутениевые комплексы экстрагируются при обработке смеси, еще дальше превращаются в другие рутениевые комплексы, и те очень часто успевают наломать дров, превратив продукты метатезиса в дымящиеся развалины тогда, когда этого уже никто не ждет. Вывод простой – надо рутений быстро вывести из реакции, причем так, чтобы он не сохранил карбеновый лиганд, который может продолжить метатезис уже там, где это совсем никому не нужно – мы же знаем, что метатезис бесконечен. Несколько решений этой задачи предложил сам Граббс, но самую быструю и удобную сделали Дайвер и сотрудники (Galan, B. R., Kalbarczyk, K. P., Szczepankiewicz, S., Keister, J. B., Diver, S. T. Org. Lett. 2007, 9, 1203) – добавлять соль изоциануксусной кислоты. Рутений так любит изоцианиды как лиганды, что моментально с ними связывается, и даже карбен меняет – причём если в катализаторе есть NHC-лиганд, этот карбен внедряется в одно из ароматических колец. В любом случае, а этот прием работает со всеми основными Ru-катализаторами метатезиса, образующиеся комплексы координационно насыщены, ни к кому не пристают, и представляют собой соль – могут быть легко отмыт водой, отделены фильтрованием через силикагель и т.п.
Блокировка смещения двойной связи
Хотя мы и вообще все все время вопим, что метатезис это высокоселективная реакция и в ней не бывает такой болезни многих реакций, катализируемых комплексами переходных металлов, как путешествия двойных связей по всем доступным им цепям, это так только для катализаторов Шрока. Но их больше никто не использует, все подсели на граббсов, а с граббсами это вообще-то неверно – гуляют в них двойные связи, ещё как гуляют. Вот например, из статьи самого Граббса (Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160). Вот сюрприз! Да тут вообще почти нужного продукта нет.
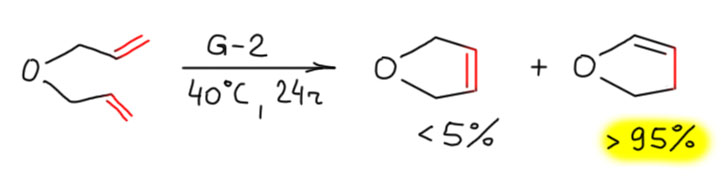
Мы, конечно, можем спросить – а зачем в такой простой реакции крутить смесь целые сутки, да ещё при нагревании? Действительно, если бы реакцию погасили раньше, то всё бы было нормально, и вообще – это же одна из самых популярных моделей для исследования RCM. Верно, но эту модель здесь и использовали, чтбы показать, что даже такая простая реакция, если ее делать неряшливо, может принести сюрпризы.
Граббс предположил, что причной проблем являются случайно образовавшиеся гидридные комплексы – но это самое очевидное объяснение блужданиям двойной связи. Осталось найти, откуда гидридные комплексы могли взяться. Нашли, получилась очень интересная работа. Чуть ниже мы ее разберем, но сначала выясним, какое лечение прописал Граббс от этого недуга. Раз там безобразничает гидрид, решил Граббс, значит надо его как-то перехватить и обезвредить. Попробовав много разных добавок, Граббс с сотрудниками остановились на бензохиноне в каталитических количествах, обычно добавляют в два раза больше загрузки катализатора; если катализатора 5 моль%, то хинона десять, и так далее. Хинон не реагирует в самом метатезисе, как и вообще электронодефицитные олефины, но вполне может реагировать с гидридным комплексом. Впрочем, мы сейчас посмотрим механизм образования этого гидридного комплекса и поймем, что бензохинон может вступить в игру раньше – там будут участвовать фосфин и илид Виттига, и то, и другое отлично реагирует с хиноном, так что до гидридного комплекса дело не дойдёт. И действительно, это оказалось эффективно, причем для всех рутениевых катализаторов метатезиса. Теперь поглядим на механизм.
Граббс с сотрудниками принялся искать, откуда мог бы взяться гидридный комплекс (Hong, S. H.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2004, 126, 7414). Подозрение пало на ту форму каталитически активных комплексов, которая содержит незамещённый карбен, метилен, и как мы знаем, эта форма – основное состояние рутениевых частиц в большинстве практических реакций метатезиса, потому что почти всегда в метатезис вводят концевые олефины. В отличие от исходной формы катализатора с замещённым, часто стерически сильно затруднённым карбеном, метиленовый комплекс намного менее устойчив, а точнее, намного более реакционноспособен. Поскольку это не карбен Шрока, такой карбен электрофилен и может реагировать с нуклеофилами. С какими? Граббс предположил, что дело в фосфине – ведь катализаторы Граббса, первый и второй, работают через спонтанную диссоциацию одного фосфина, освобождающего место в координационной сфере. Решили проверить и сделали такой карбен, и засунули его в ЯМР-спектрометр при немного повышенной температуре. И вот – комплекс и правда стал првращаться во что-то другое, и через некоторое время это другое удалось даже выделить и определить структуру. Структура оказалась просто потрясающей – это димерный комплекс с двумя атомами рутения, на одном и правда гидрид, а вот мостик – мостик оказался просто одним атомом углерода – там даже водорода нет, не потому что их не обозначили или в рентгене не увидели, нет, их там нет. Их там нет! Ха-ха, знаем мы этот нехитрый фокус! Да нет, правда нет, нет там водородов, это один углерод, один углерод – это следующая стадия после карбена и карбина.
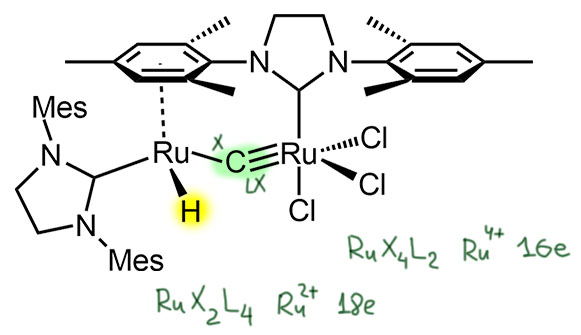
Называется карбон, вроде точно так же как углерод по-английски, но ударение не там, и если изобразить правильное произвошение, то есть разница. Это просто атом углерода, в некотором смысле еще одна аллотропная модификация: много их, от алмаза и графита до нанотрубок, графена, фуллеренов, одномерной цепи – а это одноатомный. Такой углерод в свободном виде может быть только в высоком вакууме, но современная химия вполне уже освоилась с этой формой и даже изучает ее реакции. У карбона может быть несколько форм, так же как у карбена и карбина. И это может быть лигандом, и тоже нескольких форм. Здесь проще всего вспомнить, что мы выяснили про карбины, представить себе карбин с одной стороны, причём не-Шрока, типа XL, и просто сигма-лиганд с другой стороны, чисто X. Так и образуется мостик, причем несимметричный: X и XL. И образуется очень легко.
Граббс нарисовал очень интересный механизм и частично подтвердил его. Я бы даже сказал, что мы оказались обязаны побочной реакции и получили работу, которая едва ли не интереснее собственно метатезиса и его проблем. Начинается механизм с диссоциации фосфина, но это обязательная стадия и для самого меатезиса. Можно сказать, что там где есть граббсы первый или второй, есть и концентрация трициклогексилфосфина в растворе. Проблема в том, что если карбен незамещенный, то это неэкранированный стерикой электрофил, и фосфин ожидаемо должен присоединиться. При этом меняется природа лиганда, он становиься X и на металле возникает формальный минус. Рассмотрев этот лиганд получше, замечаем, что это просто илид фосфора. Прямо как у Виттига, только с более донорным фосфином. Логично предположить, что этот лиганд может диссоциировать (это реально довольно тонкий момент, и проще было бы предположить, что уходит один из хлоридов, но ладно, Граббсу виднее). Итак, в расторе теперь плавает илид, а это не толко нуклеофил, но и нехилое основание. Но кроме илида там еще и очень ненасыщенный комплекс рутения – ободрали как липку, 12 электронов всего, вы чё, сдурели? И этот комплекс – чего мелочиться – видит мезитильную группу на лиганде SIMes у другого комплекса с еще никуда не ушедшим карбеном. Рутений цепляет эту шляпу – сразу 6 электронов и вообще, всегда бы так, только что был ободранный, а уже сразу насыщенный. Но чтобы и тому комплексу, у которого позаимствовали шляпу было не так обидно – он ведь тоже 14-электронный – ему дают два хлорида на мостики, но как L. Всё, вот вам димерный комплекс, оба рутения насыщены – такому бы жить да поживать, и даже больше лигандов не надо наживать, но плавает тут гле-то илид, про которого все забыли, а он, мятежный, ищет, как и положено сильному основанию, у кого бы оторвать протон. И находит – мы ведь уже обсудили, откуда берутся карбины не-Шрока – от карбенов надо забрать электрофуг, тот же протон, только при этом скомпенсировав заряд на металле анионным лигандом – но он-то уже есть, даже два, так что один моно вывести у металла. Получился карбин не-Шрока. И на последней стадии происходит не такая понятная перегруппировка, которая похожа на окислительное присоединение С-H к другому рутению. Одна проблема, для этого рутений должен иметь степень окисления 0, а здесь оба 2+. Тогда, возможно, это никакое не окислительное присоединение, а такое внутрисферное депротонирование карбина с репротонированием металла – это последнее не так дико как кажется, если мы вспомним, что гидридные комплексы металлов иногда бывают кислотами Бренстеда-Лоури и могут как терять протон, так и забирать его обратно именно как протон. Всё, тогда всё сошлось, механизм, прямо скажем, сказочно завирален и тем красив. Гидридный комплекс ведь получается – это доказано экспериментально. Если сможете нарисовать механизм без таких допусков – вперёд, Граббс только уже не оценит, здесь не оценит, а там может и оценит.
метатезис алкинов
DU
Очередной пример применения метатезиса алкинов для циклизации (RCAM – ring closure alkyne metathesis) в синтезе природных соединений. RCAM в отличие от RCM пригоден только для макроциклизации, но макроциклов в Природе немало, и метод оказался очень хорош, только сначала его пришлось как следует усовершенствовать и развить, чем и занялся главный энтузиаст этого метода швейцарский химик Алоиз Фюрстнер, работающий в Германии в Институте Угля в Мюльхайме. Проблема метатезиса алкинов в том, что хотя Шрок и сделал первые эффективные катализаторы этого метода даже раньше своих знаменитых катализаторов для метатезиса алкенов, дальше метатезис алкинов сильно забуксовал и катастрофически отстал от олефинового метатезиса, вызвавшего просто золотую лихорадку в синтезе. Но причиной такого невероятного успеха метатезиса алкенов стали рутениевые катализаторы Граббса – с их появлением метатезис стал совершенно рутинной химией, Метатезис мог сделать любой, кто смог раскошелиться на некоторое количество двух-трёх главных катализаторов Граббса. В метатезисе алкинов не удалось до сих пор уйти от катализаторов типа Шрока, на ранних переходных металлах – молибдене, вольфраме, рении, – а эти катализаторы требуют гораздо более умелой работы синтетика, кроме того, их больше чем 2-3, и каждую реакцию приходилось оптимизировать весьма трудоёмкими и аккуратными экспериментами. Некоторые катализаторы и предкатализаторы алкинового метатезиса н ето что с водой или кислородом взаимодействуют, но и азот не прочь порвать на атомы. Было и еще немало проблем. Вообще, если обычная циклизация на 5-6 членные циклы возникает в синтезе часто, то макроциклизация – штука редкая, и даже одно это делает олефиновый и алкиновый метатезисы несопоставимыми по востребованности. И вот Фюрстнер в конце прошлого века берётся за дело основательно, решив, во-первых, найти новые поколения катализаторов, хотя бы частично устраняющие проблемы первого поколения. И во-вторых, отладить экспериментальные методы и найти красивые применения. Во многом, хоть и не во всём, его работа увенчалась успехами, хотя алкиновый метатезис так и не стал конкурентом олефиновому – занимаются им до сих пор немногие крутые синтетики.
В данной работе Фюрстнер с сотрудниками берётся синтезировать несколько алкалоидов одной морской губки. Я уже много раз замечал, что за самыми хитроумными структурами нужно идти к самым примитивным с точки зрения Эволюции организмам – они давно живут на нашей планете, времени у них было полно, а жизнь у них довольно скучная – сиди себе где-то на дне, фильтруй воду, да задумывай самый изощрённый синтез. Несколько макроциклов – пожалуйста, тройная связь в макроцикле – пожалуйста, и т.д. Ну и Фюрстнеру и его сотрудникам забава. Зачем ты, о Губка с красивым именем Реньера из прибрежных вод Танзании, сотни миллионов лет ваяла это сооружение? – Буль-буль, чтобы Фюрстнеру было чем заняться! Ну и ещё, чтобы сделать так, что тот, кто меня, губку, по недоразумению сожрёт, пожалел бы об этом, буль-буль.
Фюрстнер мастерски собрал это сооружение и ещё несколько похожих, применив на важных стадиях метатезис алкинов. Вот, на последних стадий синтеза одного из таких алкалоидов, нджаоамина, применили RCAM. Смотрим, сначала просто, взяли некий предкатализатор, загрузочка нехилая – 30 моль%, применено две каталитические системы, лучшая дала количественный выход, итого TON всего три цикла, забавно, что время реакции довольно маленькое – реакция быстрая, проходит всего за 20 минут – TOF – один цикл приблизительно за 7 минут, округляем – за десять минут.
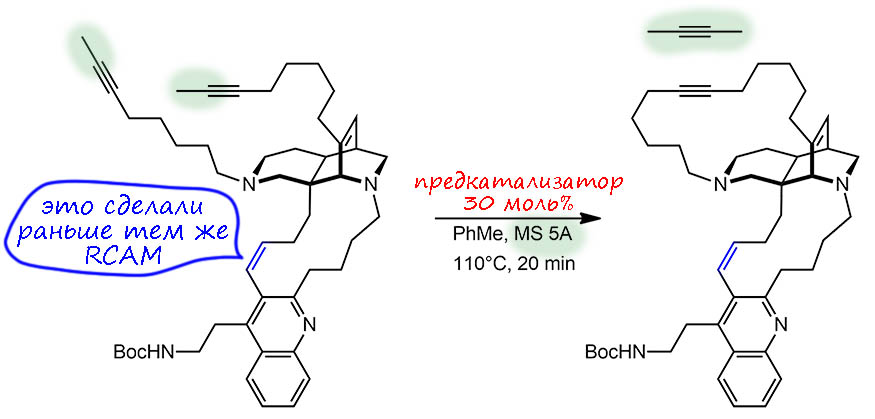
Посмотрим еще на реакцию. Во-первых, еще раз отмечаем, что в алкиновом метатезисе не используют терминальные алкины. Мы привыкли, что в олефиновом метатезисе как раз именно их и используют, потому что получающийся этилен улетает, смещая равновесие. Увы, с алкинами это не работает – терминальные алкины плохо работают в метатезисе, обычно происходит полимеризация или дело заканчивается на довольно инертном металлациклобутадиене. Поэтому приходится использовать минимальный нетерминальный алкин и тогда вторым продуктом становится бутин-2, а это хоть и газ, но тяжёлый и совершенно не торопящийся никуда улетать – в результате дело затягивается на холостые циклы. Фюрстнер раньше придумал такую уловку – добавление молекулярных сит с широкими порами. Обычно используют сита 4А, которые хватают воду и другие маленькие молекулы. Здесь же взяли цеолит с порами, способными схватить и удержать молекулу бутина – видимо, она туда втыкается метилом и так и торчит из дырок на поверхности, ну, в общем, раз получается, значит как-то помещается. Меня немного такие фокусы удивляют – ведь предполагается, что удалить из реакционной смеси нужно весь или значительную часть образующегося бутина-2, и как-то слабо верится, что у цеолита такая большая сорбционная ёмкость. Когда возникают такие сомнения, надо посмотреть эксперимент и реальные загрузки – о, там всего 7 миллиграмм исходного, огромной молекулы с молярной массой более 700, значит бутина там получится полмиллиграмма, а сит взяли 200 мг – конечно, при таком соотношении сит хватит. Но представьте теперь, что вам нужно сделать тот же метатезис с каким-нибудь алкином попроще, но загрузку взять посерьёзнее – ну, хоть несколько грамм. И сит тогда понадобится несколько сот грамм – понятно, что в химии вечно возникают такие странные затыки, что вроде на бумаге всё красиво, а начинаешь считать загрузки и получается нечто ужасное, ищем ведро, в тару поменьше не поместится. И заодно видим, что в алкиновом метатезисе, даже с уловками от Фюрстнера есть чисто практические проблемы. Но если обходиться без сит, приходится иногда реакцию останавливать, охлаждать и откачивать на вакуумной установке, чтобы удалить бутин.
Теперь про катализаторы. Одно из важных достижений Фюрстнера – введение нового поколения молибденовых катализаторов, содержащих силанолятные группы. Во всех катализаторах Шрока, что алкеновых, что алкиновых всегда долго ищут баланс между реакционной способностью и избирательностью. Катализаторы Шрока ближе всего подходят к тому, что называется катализаторами определенной структуры, well-defined catalysts – молекулы уже полностью собраны, все лиганды кроме карбина (карбена) остаются на месте во время катализа, меняется только карбин (карбен). Поэтому молекулу важно собрать так,чтобы она хорошо работала на целевую реакцию, но не приставала к прохожим. А тут и есть затык, который в метатезисе алкенов фактически катализаторы Шрока и похоронил – либо у вас катализатор активен, и тогда он должен иметь координационную ненасыщенность и достаточно места в координационной сфере (она нужна для связывания алкена или алкина через дигапто-комплекс, предшествующий образованию металлацикла), достаточную электрофильность (катализаторы Шрока заведомо работают через электрофильный режим активации кратных связей, просто по определению, ведь они все содержат металл в максимальной степени окисления, кислоту Льюиса) – но та же реакционная способность заставляет катализатор легко ловить донорные группы с кислородами или азотами, и тогда такие группы могут блокировать катализ. Так же действует влага или донорные растворители – всё это приходится избегать, вести реакции в тщательно высушенных системах – не беда, но создаёт дополнительные неудобства. А карбеновая или карбиновая части наоборот нуклеофильны, и тут тоже легко получить проблемы, например, с карбонильными группами.
Иными словами, дизайн катализаторов типа Шрока для метатезиса – кропотливый поиск баланса между реакционной способностью (она же каталитическая активность) и оптимальной избирательностью, она же толерантность к заместителям и функциональным группам. Мы знаем, что в алкеновом метатезисе поиск баланса привел к необходимости иметь алкоксильные группы на металле, и далее их ещё тюнинговали фторированием. Но пришёл Грабс – и выгнал всех к чёртовой бабушке. В алкиновом метезисе начинали с того же, и главным катализатором долго был карбиновый комплекс вольфрама с теми же фторированными алкоксидами. Вообще, большинство катализаторов алкинового метатезиса используют металлы 6-й группы, кроме хрома, и у них одинаковый тип -карбин и три X-лиганда как раз для того самого тюнинга.
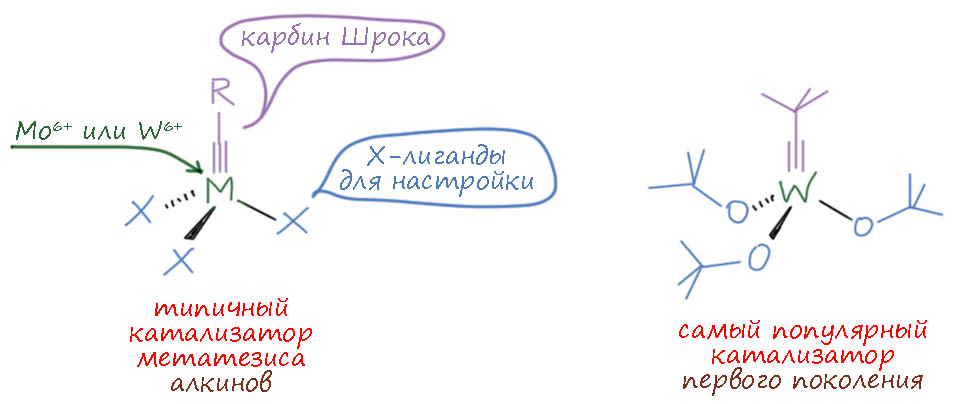
У типичного катализатора алкинового метатезиса полно уязвимостей – можно сказать, что это такая заведомая развалина, – там болит, здесь болит… Карбиновый углерод боится электрофилов, напрмер, кислот, даже слабых, типа малонового эфира. Металл только и думает о том, как бы подцепить ещё какую-нибудь донорную дрянь, к счастью, это часто обратимо, но в любом случае требует повышенных температур, чтобы равновесие было смещено в сторону диссоциации. Ещё хуже с лигандами, которые долго и кропотливо подбирали для достижения нужного баланса – любой нуклеофил, особенно в условиях общегл кислотного катализа, так и норовит снести и сесть самому на это место, куда его никто не звал, и где он необратимо разрушит деликатный баланс. Поэтому метатезис становится проблемой, если в субстрате есть заместители с такими свойствами и реакционной способностью – как минимум TON становится неотличим от единицы, как максимум вместо желаемого продукта получается – ничего не получается.
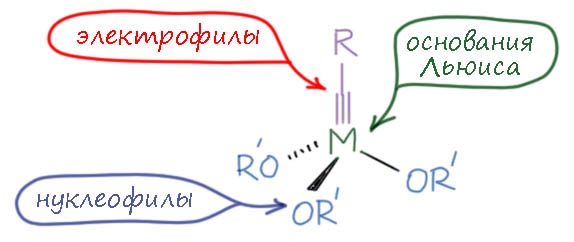
Фюрстнер с сотрудниками сначала постарались заняться верхом этой картинки – настройкой электронных требований металла и карбина. Эти поиски привели их к тому, что вместо алкоксильных надо использовать кремниевые аналоги. Стоит понимать, что все такие исследования основаны на 99% на кропотливой экспериментальной работе – исследователи типа Фюрстнера, работающие в очень крупных и богатых исследовательских центрах и не испытывающие проблем ни с финансированием, ни с обеспечением, ни с желающими поработать, могут себе позволить делать огромные объёмы экспериментальной работы – и когда что-то начинает получаться, приходит очередь красивых обоснований и работе на то, чтобы доказать, что найденное является результатом могучей работы мысли. В химии 21 века практически так же как в химии 19 века на первом месте эксперимент, на втором везение, и только на третьем (а точнее, на десятом, но тогда непонятно, кто занимает места с 3-го по 9-е) теория. Вот и Фюрстнер обосновывает открытие нового поколения катализаторов изощренными доводами, например тем, что кремниевые аналоги алкоксилатов не просто менее донорны, но и якобы умеют подстраивать донорность в зависимости от ситуации – то увеличивая электрофильность молибдена (а катализаторы этого поколения все молибденовые, вольфрам и рений пока отодвинули), то уменьшая её, потому что на стадии связывания алкина нужно одно, на стадии циклореверсии другое. Может это и так, но выглядит довольно наивно. Катализатор впрочем оказался работающим и действительно справлялся со сложными субстратами, имеющими донорные атомы кислорода или азота (но не свободные спиртовые группы). Катализатор можно готовить загодя, хотя его приходится стабилизировать дополнительными донорными лигандами, и дальше в реакции реактивировать комплекс. А можно, как в рассматриваемой статье брать предкатализатор, амидный комплекс молибдена и силанол. С просто силанолами Фюрстнер играл ещё 10 лет назад, получалось неплохо, но такие лиганды оказывались слишком лабильными, и тогда было решено противодействовать лабильности хелатированием – придумали тридентатный лиганд (его еще называют триподальным – то есть “с тремя ножками”) – получилось лучше. Вот ровно такой катализатор и используют в рассматриваемой работе, причем не делают его заранее, а добавляют молибденовый амидный комплекс и лиганд – амидные лиганды быстро меняются на силанольные благодаря очень высокой основности.
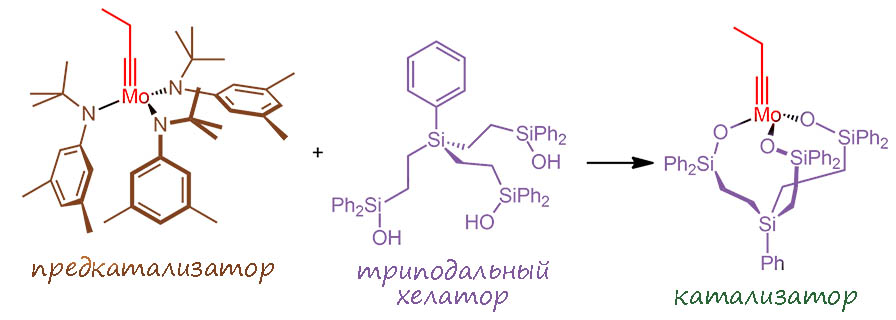
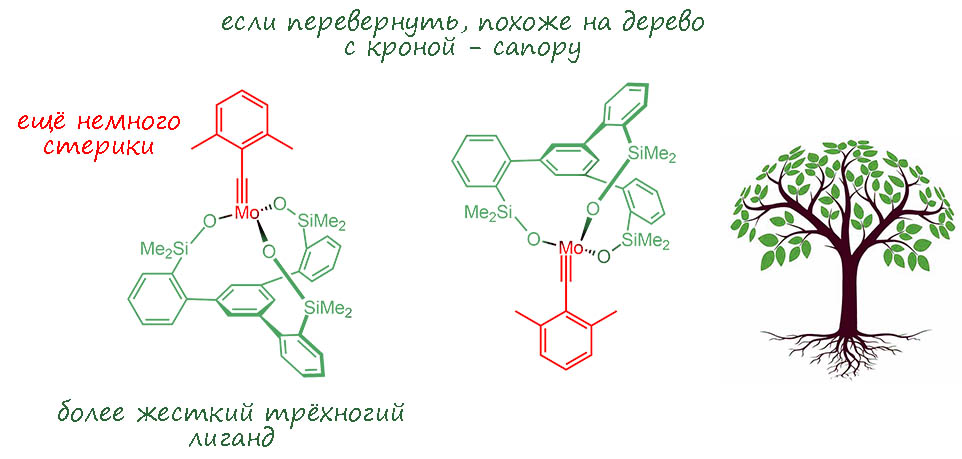
Реакции прошли (а в работе не только рассматриваемая реакция, но и еще несколько метатезисов) успешно, продукт получился, хотя и с немаленькой загрузкой предкатализатора – получился TON около двух – два каталитических цикла. Фюрстнер стал искать дальнейшего улучшения, решив, что такой триподальный лиганд слишком гибок, и хелат получается непрочный, более того, образуется далеко не количественно, и быстро деактивируется. И совсем недавно прдложил очередное улучшение, названное canopy catalyst – от слова, означающего купол, навес, крона дерева (Hillenbrand J., Leutzsch M., Yiannakas E., Gordon C. P., Wille C., Nöthling N., Copéret C., Fürstner A. J. Am. Chem. Soc. 2020, 142, 11279−11294). В общем, то же самое, но лиганд взяли более жёсткий, и более того, имеющий конформацию с тремя сближенными и скрепленными водородными связями гидроксилами, поэтому он очень быстро и количественно связывается к комплекс. Вот этот катализатор показал выдающиеся свойства и отлично работал в метатезисе алкинов, содержащих всевозможные функции, в том числе свободные гидроксилы и т.п. В этой работе такой катализатор и дал почти количественный выход. правда тоже с большой загрузкой и маленьким TON.
Карбиновые лиганды: что это такое?
Раз мы сюда забрались, у нас появился отличный повод и даже инструмент немного лучше разобраться с карбиновыми комплексами. В первой части квеста было три таких. Что с ними делать? Что такое вообще карбиновый лиганд?
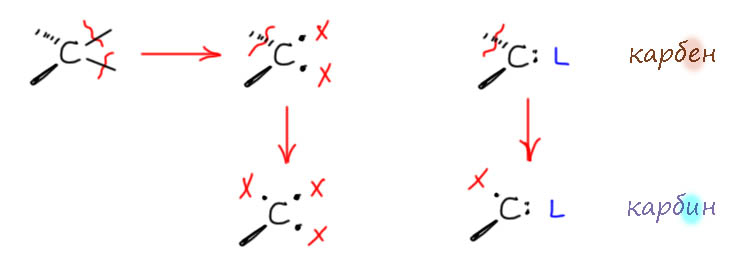
Есть очень большой соблазн сказать, что это что-то типа карбенового. С карбенами разобрались, значит и с карбиновыми разберёмся. Но сразу возникает маленький вопрос: карбены бывают двух типов – Шрока и Фишера в обобщённом смысле. Мы договорились, что карбены Фишера в специальном смысле это нечто особенное, особый тип комплекса, но довольно часто в литературе все карбеновые лиганды типа L называют карбенами Фишера, считая это таким антонимом к термину карбен Шрока, то есть лиганд типа X2. Мне очень не нравится эта неразборчивость и путаница и я не рекомендую эту обобщённую трактовку термина “карбен Фишера”, но против устояшейся традиции трудно переть. Хорошо, хоть так, хоть так, смысл понятен, – карбеновых лигандов есть два типа – и теперь посмотрим, что там у карбинов.
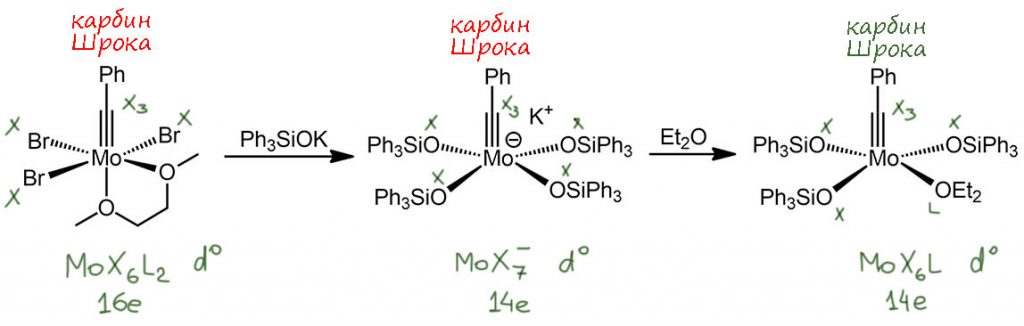
Проще всего с карбинами Шрока, которые сам Шрок не велел называть карбинами, а велел называть алкилидинами. При всём почтении к выдающемуся ученому, мы не послушаемся, тем более, что он и свои карбены не велел называть карбенами, а велел – алкилиденами. А все называют, правда, добавляя – Шрока. Поэтому и мы назовем карбины карбинами Шрока. Но тут же встаёт вопрос – а все карбины таковы? Карбины Шрока мы будем искать там же, где и все такие ситуации, когда back-donation заходит настолько далеко, что превращается в полноценную связь, а природа лиганда изменяется с L на X2 – практически все такие случаи у ранних переходных металлов (а это всё вплоть до 7-й группы) проявляются тогда, когда металл получает возможность сбагрить все валентные электроны и достичь конфигурации d0. Вот, из квеста одна структура точно подпадает под это условие – BN. Это рениевый комплекс (Fc – это ферроценил) с одновременно карбеновым и карбиновым лигандами. Если оба принять за полностью анионные, то рений и получает конфигурацию d0 – это карбен и карбин Шрока. Проблем нет.
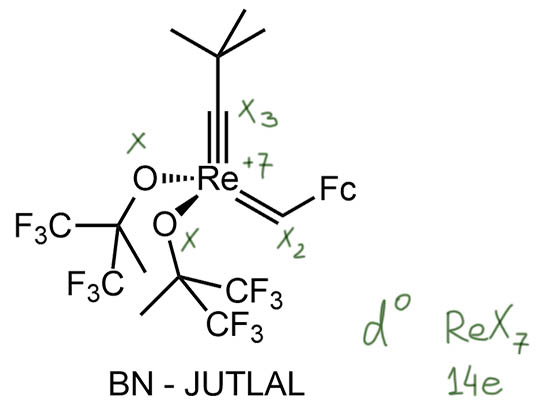
А вот с двумя другими не все так ясно. Они одинакового типа, поэтому посмотрим на более простой комплекс хрома. Если карбин принять за X3, у хрома будет степень окисления +2 и конфигурация d2 – не то, чо хотелось бы. Но это же не закон Природы, а просто тенденция, почему бы ей иногда не нарушаться. Да, никаких препятствий нет, и так часто и делают – принимают карбин и здесь за карбин Шрока, трианионный лиганд, тем более что степень окисления +2 для 6-й группы вполне часто встречается, и эти электроны исподтишка растащат карбонилы, но мы знаем, что карбонилы не принято считать чем-то иным кроме L, просто такая конвенция, ничего личного.

А может быть другая интерпретация? Какая же? Ну, например, тоже L? Но тогда карбин будет неотличим от карбена, а это точно не годится. Да и степень окисления хрома в этом комплексе будет +1, а это уже очень большая редкость и неспаренный электрон и парамагнитность – но ничего подобного про такие комплексы неизвестно, они диамагнитны. И ещё одно непонятно – почему в таких комплексах всё время на металле есть один явный X-лиганд, обычно галоген, ведь если бы его не было, гипотеза о том, что карбин – нейтральный L-лиганд, просто не с одной, а с двумя p-орбиталями для back-donation, могла бы прокатить, как минимум для затравки.
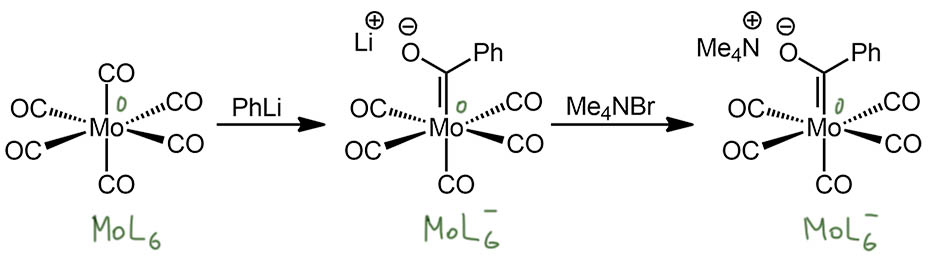
Интересно, что когда мы сталкиваемся с не совсем понятными комплексами и не можем легко решить, как связан металл и лиганд, есть отличный способ разобраться – посмотреть, как этот комплекс получается. Разберем путь синтеза из работы Фишера, после в разных вариантах многократно использовавшийся. Итак, мы начинаем с гексакарбонила, например, молибдена. Сначала действуем так, как мы уже разбирали – присоединяем литийорганику, получаем анионный комплекс, но с зарядом на лиганде, а тип комплекса и степень окисления не изменяются. Образовавшийся лиганд это карбен, в комплексе – карбен Фишера, что мы и обозначили двойной связью, показав back-donation. Замена противоиона нам ничего не даёт, но это приём, который использован для очистки комплекса.
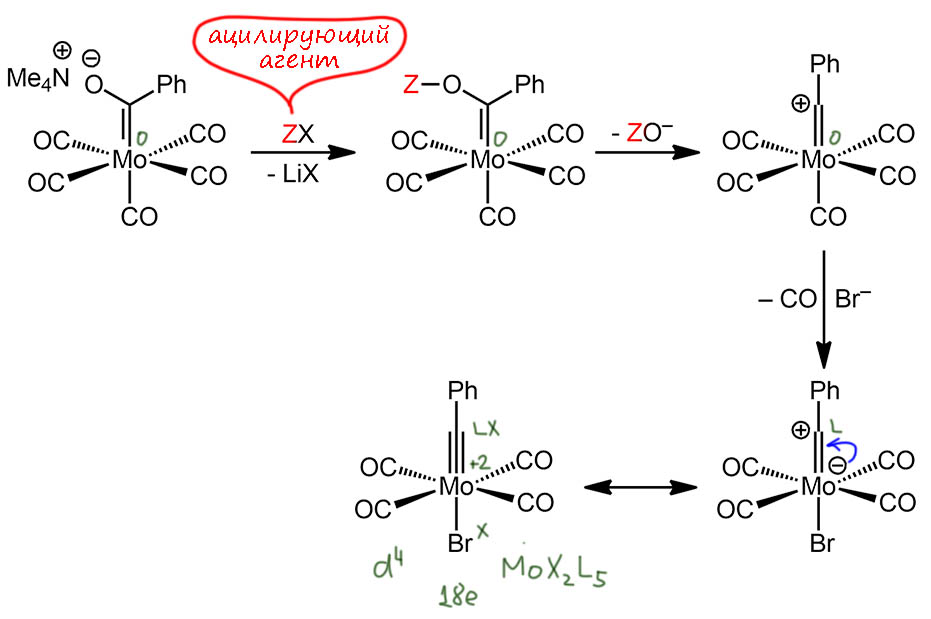
Что произошло дальше посмотрим сначала в общем виде. Кислород можно ацилировать или сульфонилировать, и тогда мы получаем уходящую группу. И если она уйдёт, то образуется фактически катион, но рядом металл с кучей электронов, ведь до сих пор он оставался нульвалентным. И почему бы металлу не дать пару электронов, чобы заткнуть эту дыру? Но вот жадина, не даёт. Почему не даёт? Да, на нём одни карбонилы, акцепторные лиганды, они давно под шумок растащили всю электронную плотность с металла. При советской власти, будь она трижды забыта, такое называлось растратой. Стоит склад, на складе по ведомости хранятся всякие ценные вещи, капуста, например. Приходит проверка, такое специальное ведомсво было, ОБХСС – отдел борьбы с хищениями социалистической собственности – берут за воротник директора склада – где капуста? Нет капусты, сгнила, плохая была – так, а откуда жигули на складском дворе? 10 лет за расхищение социалистической капусты! Вот и тут – на металле написано “шесть электронов” – а для карбокатиона-лиганда жалкой пары не находится, и карбонилы довольные лоснятся от чужой электронной плотности. И при этом нам не разрешают никак этого обозначить – первое правило координационной химии: карбонил всегда L-лиганд, хоть тресни! Мы уже сто раз выясняли почему – потому что эффект переменный, зависящий от множества факторов и ничего конкретного сказать нельзя, поэтому проще не париться, пытаясь как-то это учесть в обозначениях, но при этом иметь в виду, что если на металле куча карбонилов, то его реальная степень окисления имеет мало общего с тем, что написано. Иначе вместо простой и удобной схемы – лиганды бывают двух типов, L и X, пришлось бы вводить переменные типы, настраиваемые типы, нести какой-то вздор типа – а здесь лиганд на 41% икс, и на 59% эль, а вот, птичка пролетела, и лиганд уже на 29% икс – кому нужен такой вздор, в науке терминология должна быть определённой и достигается это всегда за счет некоторого разумного уровня абстракции. Вернёмся к комплексу: но тут происходит интересное – лигандный обмен, один карбонил меняется на бромид, а бромид отрицательн заряжен, значит и в комплекс приходит этот заряд. И вот еще одна гримаса координацонной химии: карбонил – акцептор, а галоген – донор, он принёс электроны. Хорошо – так отдайте их карбокатиону! Отдаём и тут прямо прозреваем – вот оно, оказывается, в чём дело, зачем бромид и что за лиганд карбин в общем случае – это XL-лиганд. Не в том смысле, что он большой-пребольшой, а в том, что он а) 4-электронный; б) изменяет степень окисления металла на +1. И ещё один X-лиганд, бромид, итого степень окисления металла +2, а счет электронов не изменился – полные 18 электронов.
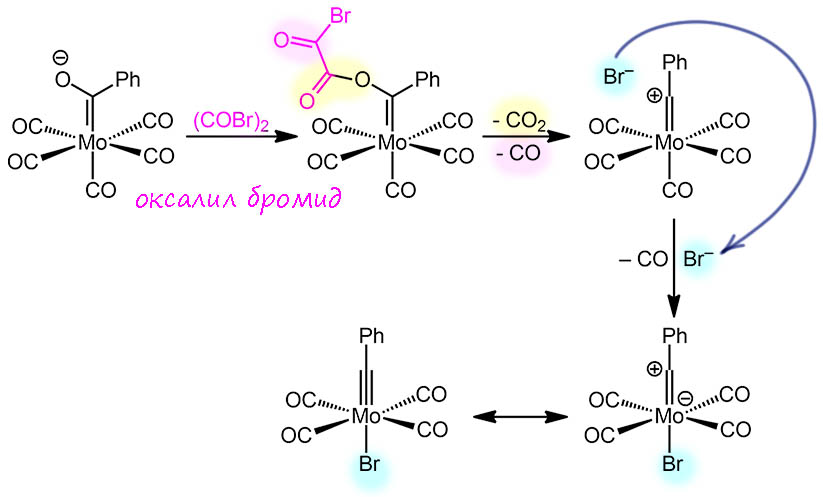
Немного допилим. Что там за ацилирующий агент, превращающийся в такую хорошую уходящую группу, ведь наверное при всём содействии металла, которое нужно ещё подтолкнуть правильным лигандным обменом, не так выгодно уйти и оставит за спиной карбокатион, да ещё такой, от которого в обычной органической химии все выпали бы в осадок. Очень просто – используется бромангидрид щавелевой кислоты, а это такая занятная штука, что её уход немедленно приводит к необратимому распаду на два оксида углерода и галогенид – поэтому это невероятно выгодно.
Итак, если карбин не Шрока (ещё раз напомню – главный признак это достижение металлом конфигурации d0 ), то лучше считать этот лиганд 4-электронным лигандом XL-типа. Карбин Шрока – 6-электронный, X3. Карбин не-Шрока вносит +1 в степень окисления металла, но всегда сопровождается еще одним X-лигандом типа галогенида, итого +2. Карбин Шрока вносит +3 в степень окисления металла. Карбины не-Шрока логично было бы называть карбинами Фишера, тем более, что именно этот великий металлорганик и получил и описал их первым, но почему-то это не очень принято. Чёрт знает почему, не исключено даже и то, что в англоязычной литературе, то есть иными словами в почти всей литературе, тоже есть всякие околонаучные тёрки, и я не исключаю, что Уилкинсон и его многочисленные ученики и последователи (сам Шрок был связан с Уилкинсоном через своего учителя Осборна, можно сказать, что он такой научный внук Уилкинсона), а это чуть ли не половина крупных имён в металлоорганике переходных металлов второй половины прошлого века, так и не смогли простить Фишеру его бесцеремонное вторжение в химию ферроцена, и он так и остался для этой тусовки чужаком, тем более что упорно продолжал писать свои статьи по-немецки в немецких журналах. Я никак это не оцениваю, ничего страшного в этом нет, даже если это действительно так, наука – дело честолюбивых и гордых людей, точно так же как искусство, бизнес, политика, и нет никаких способов сделать так, чтобы все с утра до вечера дружили, нахваливали друг друга и уступали место в истории.
К карбиновому лиганду можно подступиться даже ещё проще. Вот карбеновый лиганд – что это? Это двухвалентный углерод – то, что получится, если от четырёхвалентного оторвать два валентности. В смысле, порвать две связи – на их месте образуется по электрону, всего два электрона, и есть два варианта – оба неспаренные (это триплетный карбен, а как лиганд – это X2 потому же почему метил – это X-лиганд) или один спаренный, пара, и еще вакансия (это синглетный карбен, или L-лиганд с вакансией для принятия back-donation). Теперь карбин – это что? Это одновалентный углерод, или обрубок с разорванными тремя связями. То есть, это либо три неспаренных электрона, и как лиганд это X3 по той же логике. Или два электрона спариваем в пару, но один остается и это и есть X и L. Третьего варианта не видно. Только ни в коем случае не думайте, что это описание реальной имии – типа, “триплетный карбен вв комплексе дает карбен Шрока”. Нет, это полный вздор и более того, за исключением стабильных карбенов типа NHC, мы не получаем карбеновые комплексы из свободных карбенов, какие бы они ни были. Поэтому речь идёт только о формальном описании по валентным электронам, но это оказывается вполне полезно.
Вернёмся ещё раз к химии. Вот реакция, которая делает карбин Шрока из карбина не-Шрока. Просто обычный бром убирает карбонилы с молибдена, скорее всего, окисляя их в бромфосген, при этом карбиновый лиганд остается на месте – вот ведь химия, кто бы предсказал, что карбонил оказывается более подвержен окислению, чем карбин. Но карбин сильно электрофилен, и с электрофильным бромом не реагирует – так что всё правильно. В этой реакции L-лиганд карбонил замещается на X-лиганд бромид, поэтому металл изменяет степень окисления (был Mo(2+) стал Mo(6+)), но не только – он становится более склонным к back-donation на карбин (раньше всю эту плотность растаскивали пи-кислотные карбонилы), и появляется смысл в том, чтобы и оставшиеся два d-электрона сбагрить – и готово, вот и карбин, оставаясь тем же самым лигандом фенилэтилидином, меняет природу, становясь X3. И не смущайтесь тем, что в реакции, которая выглядит как обмен лигандов, изменяется степень окисления металла – это не обмен, это окислительная реакция – Br(0) забирает электроны, превращаясь в Br(1-).
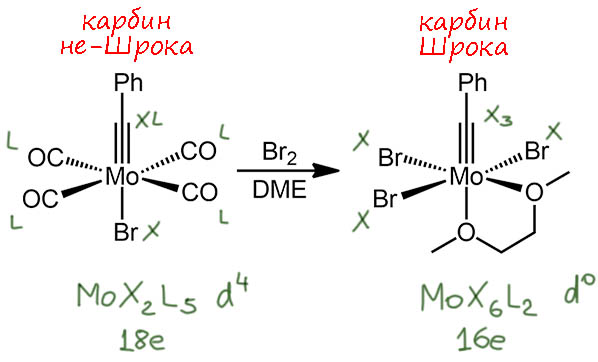
Этот путь часто применяют для синтеза эффективных пред-катализаторов алкинового метатезиса, например, Фюрстнер так получил серию высокоактивных катализаторов с силанолятными лигандами. Происходит просто лигандный обмен, авысокая электрофильность высоковалентного металла проявляется в легкости связвания донорных лигандов, впрочем, они остаются весьма лабильными и легко меняются дальше. Обратим внимание, что при замещении бромидов и диметоксиэтана (DME) уменьшается счет электронов – явное следствие увеличения стерического объёма лигандов, так что хелатный DME замещается на один, а не два лиганда. Мы в самом начале утверждали, что при замещении лигандов ни степень окисления, ни счёт электронов не изменяется – вот вам исключение, вызванное именно стерикой и больше ничем – ну не помещается там больше. А на последней стадии всё сохраняется.