Реакции с участием оксида и диоксида углерода
Углерод как элемент тверд и поэтому малопригоден для нормальных реакций, так как образует в твердой фазе бесконечные двухмерные или трехмерные цепочки прочно связанных друг с другом атомов, которые нужно сначала как-то контролируемо разрушать. Это требует огромных затрат энергии, и, соответственно, условий, трудносовместимых с нормальными реакциями синтеза. Для того, чтобы участвовать в хороших, то есть разнообразных и синтетически продуктивных реакциях углерод нужно превратить во что-то простое, но более доступное в виде отдельных молекул, на которые можно культурно воздействовать реагентами и катализаторами. Самые простые молекулярные представители элемента углерода – оксид и диоксид углерода, а также метан. Эти три соединения составляют то, что часто называют C1-химией, имея в виду то, что преобразование одноуглеродных молекул в более сложные – достойная задача органического синтеза, позволяющая воспроизводить все многообразие органических молекул из очень простой и практически неограниченной сырьевой базы. Мы отлично знаем, что с этой задачей превосходно справляются бактерии и растения, и нам как-то неудобно считать себя синтетиками низшего порядка по сравнению с ними. Про метан мы поговорим отдельно, а здесь займемся оксидами углерода.
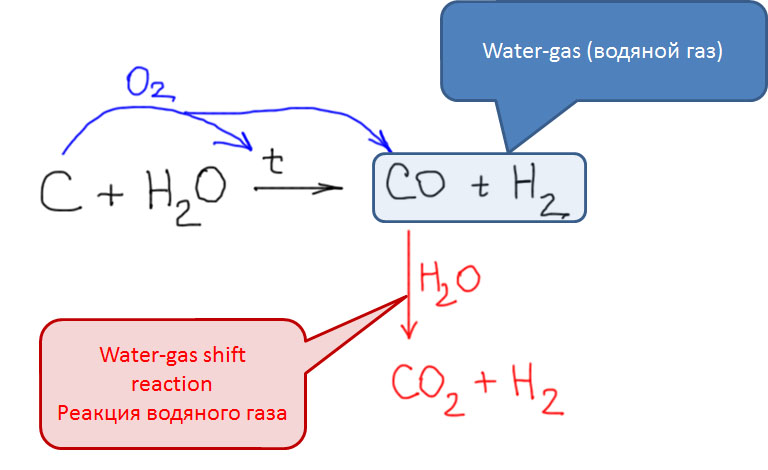
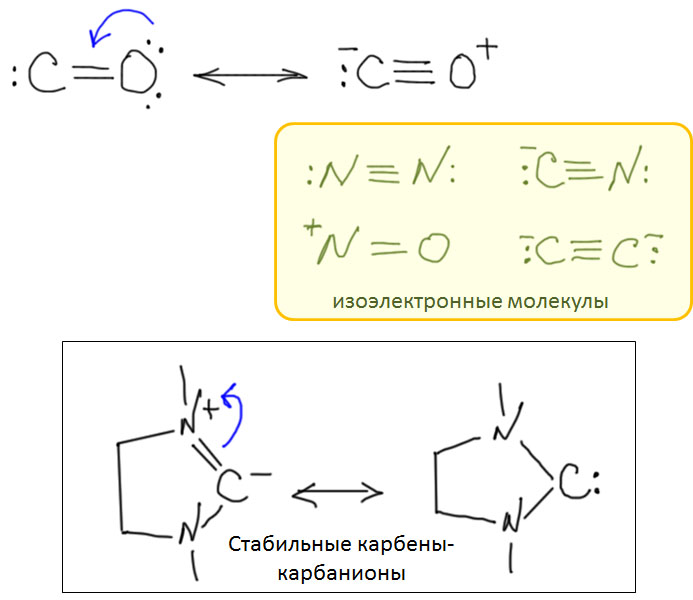
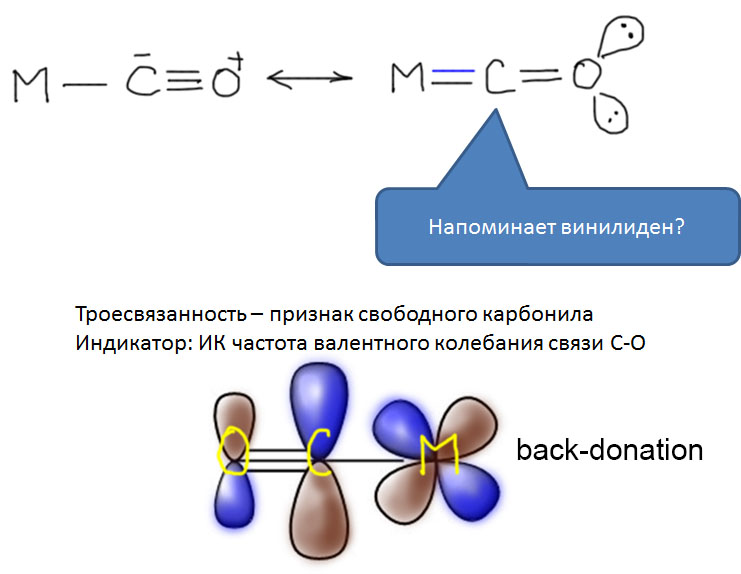
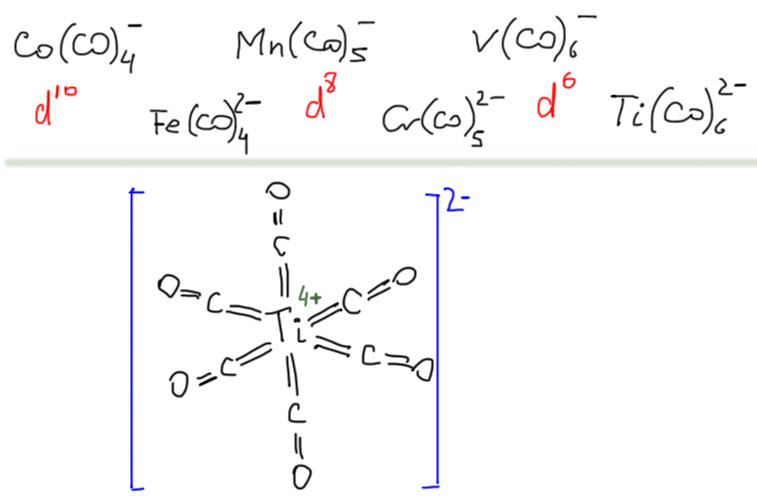
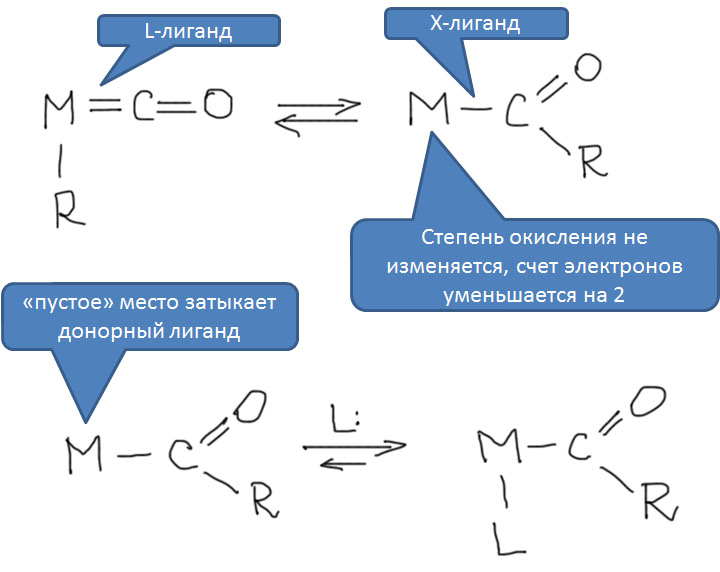
Из двух обычных оксидов углерода, монооксид CO обладает невероятно высоким авторитетом в химии переходных металлов. Оксид углерода или просто карбонил – один из самых важных и наиболее подробно исследованных лигандов, а реакции этой простой молекулы многочисленны и разнообразны. Это само по себе отлично, но есть одна проблема – оксид углерода не является непосредственно доступным производным углерода – он, к счастью, не встречается на планете Земля в ощутимых количествах, его нужно делать из угля (в промышленности), или муравьиной кислоты (в лаборатории). Это вещество весьма токсично, при этом оно не обладает ни вкусом, ни запахом, что делает его чрезвычайно опасным – люди незаметно травятся оксидом углерода насмерть. В лабораториях и промышленных помещениях, где работают с оксидом углерода, используют всякие способы распознать опасную концентрацию, от автоматических датчиков до бумажек, пропитанных солями палладия. Дурная слава незаметного убийцы, тянущаяся за этим веществом, лишает многих всякого желания встречаться с ним в работе и жизни. В лабораторных условиях существует множество способов генерировать CO прямо в реакционной смеси из более безобидных или, по крайней мере, осязаемых веществ, но дискомфорт все равно остается.
Диоксид углерода в смысле доступности и опасности отличается от CO радикально. Колоссальные количества этого вещества находятся в атмосфере и ежесекундно вырабатываются при сжигании топлива и жизнедеятельности живых организмов. Проблема глобального потепления как раз и связана с значительным повышением концентрации диоксида углерода в атмосфере по причине деятельности человека. Любители спорить с этим как-то совершенно не учитывают тот очевидный факт, что человечество за сто с небольшим лет достало из недр земли и вернуло в атмосферу то, что в этих недрах накапливалось миллионами лет, уходя как раз из атмосферы за счёт фотосинтеза и захоронения растительных остатков на больших глубинах. Тут как раз неплохо бы не доказывать, почему в атмосфере так сильно возрос уровень углекислоты, а желающим это опровергнуть – попробовать объяснить, куда подевался такой чудовищный объём возвращённого в атмосферу углекислого газа. А объём действительно чудовищный, ведь львиная доля потребностей всего человечества в энергии была покрыта за счёт сжигания ископаемого топлива.
Многим приходит в голову идея, что углекислый газ нужно из атмосферы забирать. А куда девать? Самое надёжное было бы отправить обратно в глубины земли. Но как это сделать совершенно непонятно. И поскольку человек – существо промысловатое, рождаются идеи как бы не просто углекислый газ из атмосферы забирать, но ещё что-то полезное из этого делать, что можно хорошо продать и тем самым покрыть расходы на это производство, да еще и наварить немного. Получить пока из CO2 ничего особенного не удалось, но саму эту идею удалось хорошо продать политикам разных стран и чиновникам международных организаций так, что исследования в этой области в последние 10 лет стали финансировать не просто щедро, а прямо баснословно щедро. В научных журналах последних 10 лет просто глаза рябит от статей про новые реакции из CO2, и в начале почти каждой такой статьи будет обязательно написано про благородную цель избавить человечество от медленного поджаривания и одновременное получение ценнейших материалов, лекарств, полимеров, и т.п. Большинство таких исследований производят совершенно комическое впечатление. Что-то полезное в них подчас действительно получается, но авторы умудряются не замечать двух вещей: мизерных потребностей человечества в предлагаемых продуктах и огромных затрат материалов и энергии на осуществление предлагаемой реакции. Проблема в том, что CO2 из атмосферы нужно убирать в масштабах десятков гигатонн, а всё производство органической продукции химической промышленности оценивается величинами в сто раз меньшими. Поэтому даже в совершенно фантастическом варианте, когда вообще все органические материалы будут получать из CO2, количество забранного из атмосферы на эти цели диоксида углерода было бы пренебрежимо мало и на баланс углерода серьёзного влияния бы не оказало. Точность современных оценок этого баланса гораздо больше, чем эти количества, и титанических усилий поанета наша просто не заметила бы. Но, увы, это ещё не всё. На любой химический процесс тратится очень много энергии и материалов. В расчётах все такие затраты пересчитывают на затраченную энергию, и делают простую оценку, сколько CO2 при этом будет выброшено обратно в атмосферу – это называется “углеродным следом” продукции или процесса. Проблема ведь в том, что энергию до сих пор в основном получают сжиганием ископаемого топлива. Можно просто взять энергетические и топливные балансы крупных стран, чтобы в этом убедиться. Да, когда-нибудь энергию будут получать целиком из возобновляемых источников с нулевым углеродным следом. Но тогда и выброс CO2 в атмосферу уменьшится действительно радикально, планета придёт к новому балансу, и смысла забирать из атмосферы углекислоту не будет.
Всё это безусловно не значит, что заниматься химией CO2 не стоит. Химией вообще заниматься стоит, но именно с целью получения полезных материалов и веществ. Если будет хороший процесс с использованием углекислоты – очень хорошо.
, и извлекать его оттуда было бы очень выгодно. На самом деле, это было бы так, только если бы это удавалось делать в совершенно циклопических масштабах и только в том случае, если бы эта деятельность происходила бы за счет энергии возобновляемых источников. Поскольку ни о том, ни о другом речь не идет, нам остается только удовлетвориться обычной отговоркой, что с чего-то нужно начинать. Диоксид углерода нельзя назвать совсем безопасным газом, и лишиться жизни с его помощью тоже можно, но все же и концентрация для этого потребуется на порядки более высокая, и накопление реально опасных концентраций этого газа требует особых условий – замкнутых помещений, и почти полного отсутствия газообмена. Казус известных пещер вокруг супервулкана Флегрейских полей в окрестностях Неаполя, где плохо себя чувствуют маленькие собаки, а люди даже не замечают проблем, показывает еще одно спасительное свойство этого тяжелого газа – он скапливается в опасных концентрациях только снизу. В общем, не реагент – мечта! Еще и приплачивать будут, если вы его куда-нибудь засунете. Увы, это оказывается настолько непросто, что реальных успехов в химии диоксида углерода пока что кот наплакал. Почему все так печально, посмотрим, когда доберемся до этой интересной, но немного импотентной молекулы.
Начнем с оксида углерода.
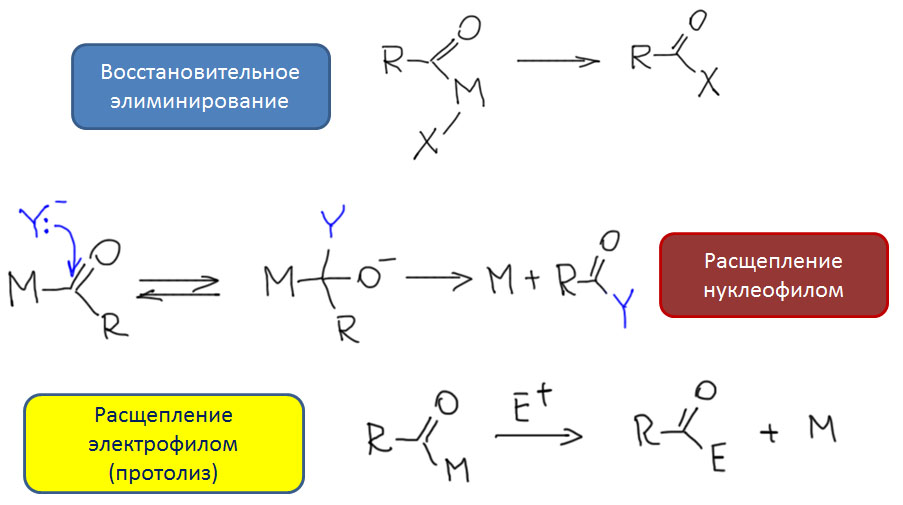
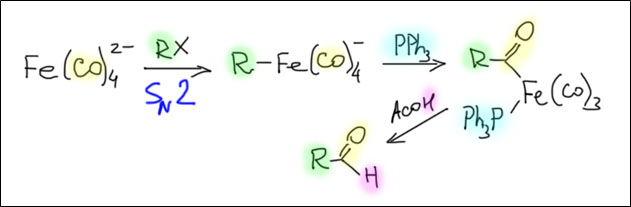
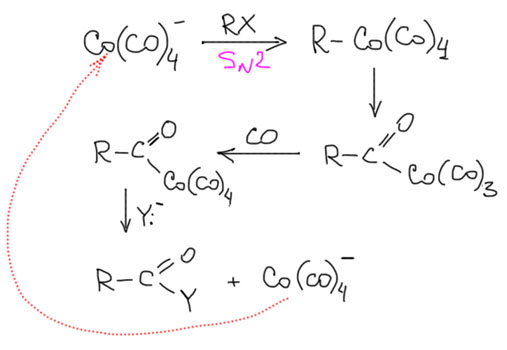
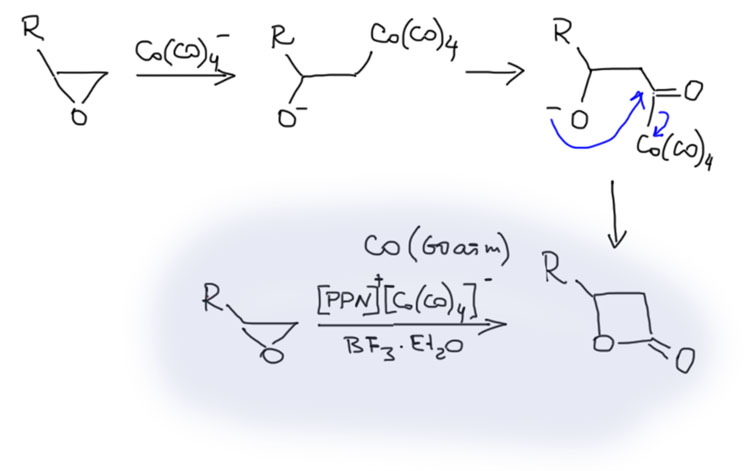
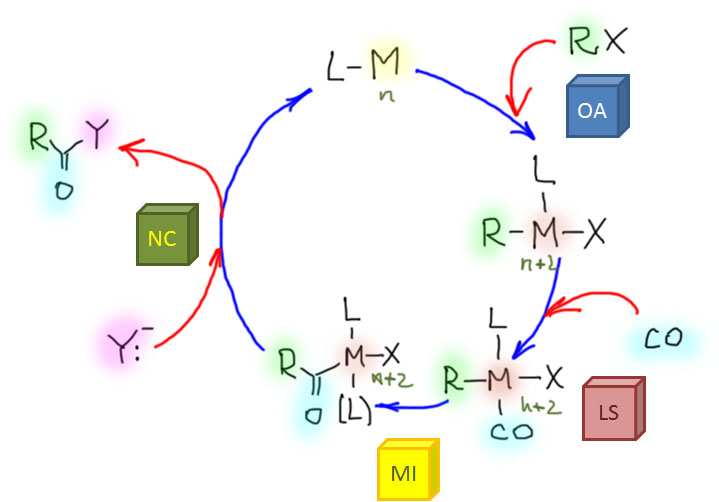
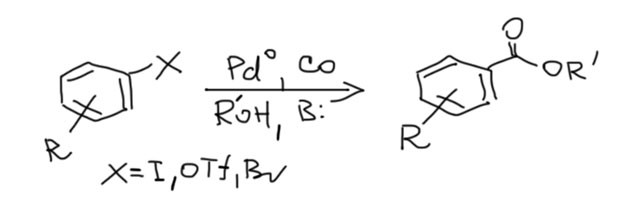
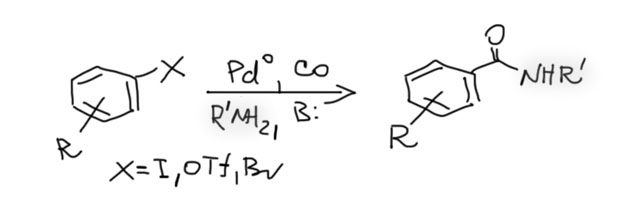
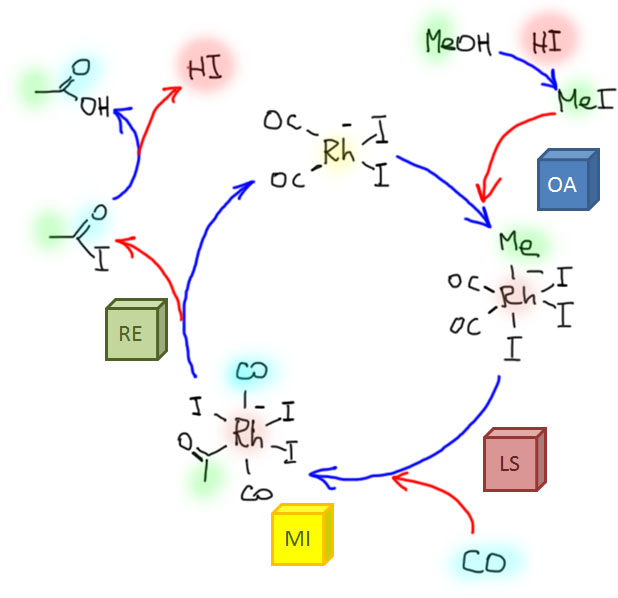
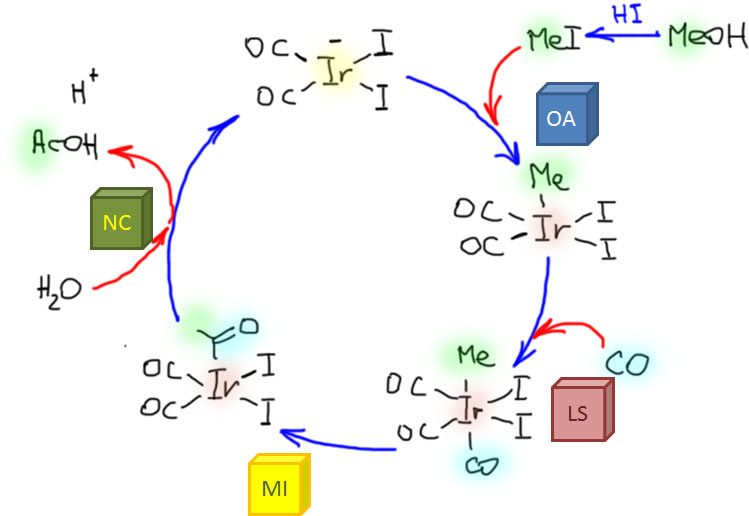
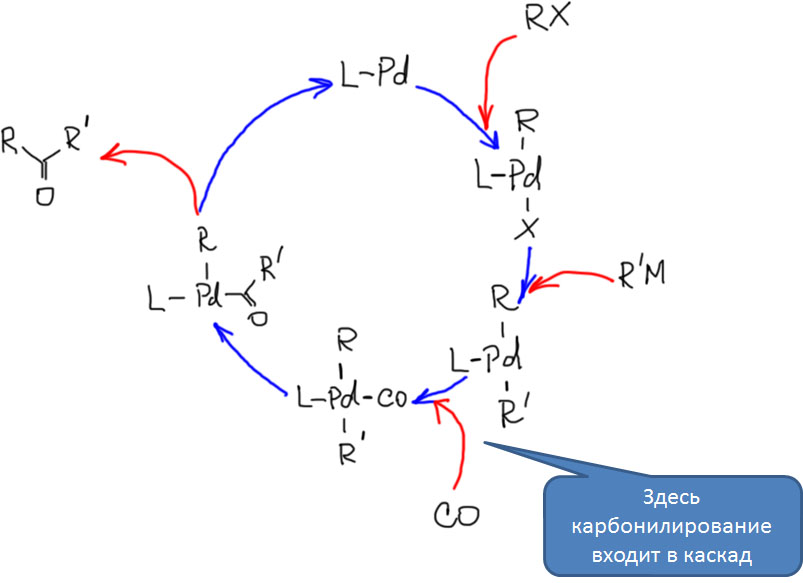
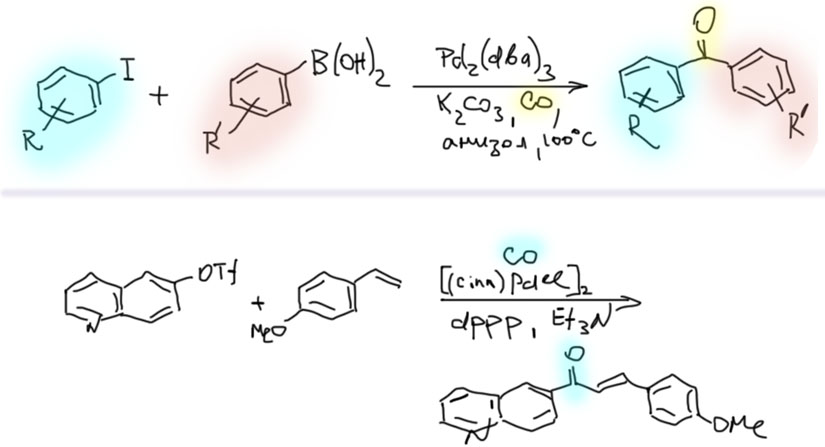
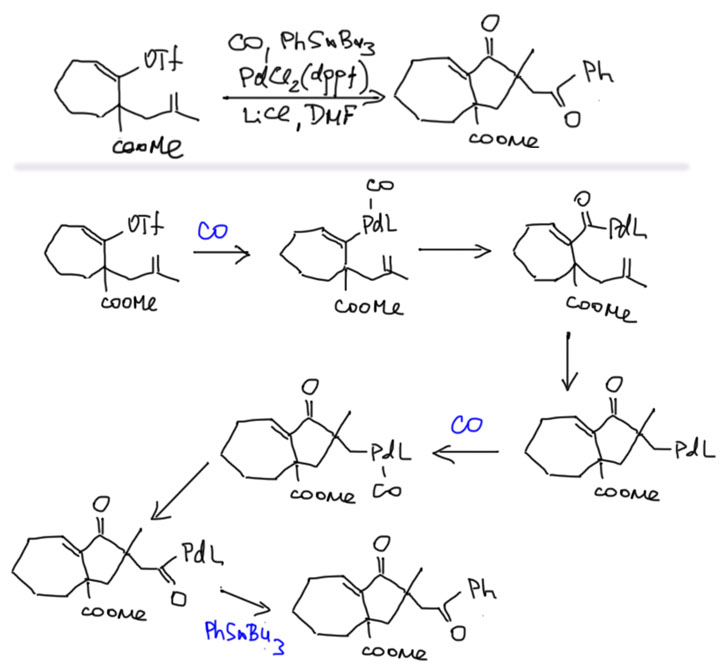
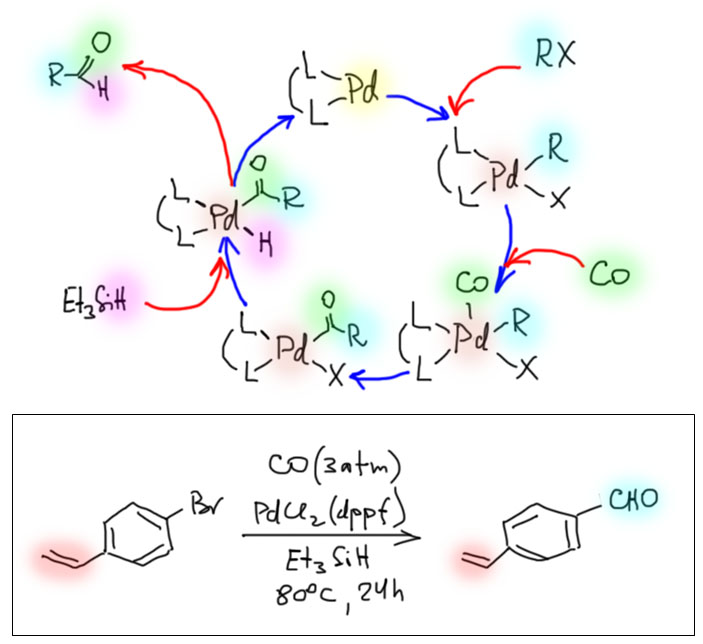
Реакции олефинов и ацетиленов с оксидом углерода
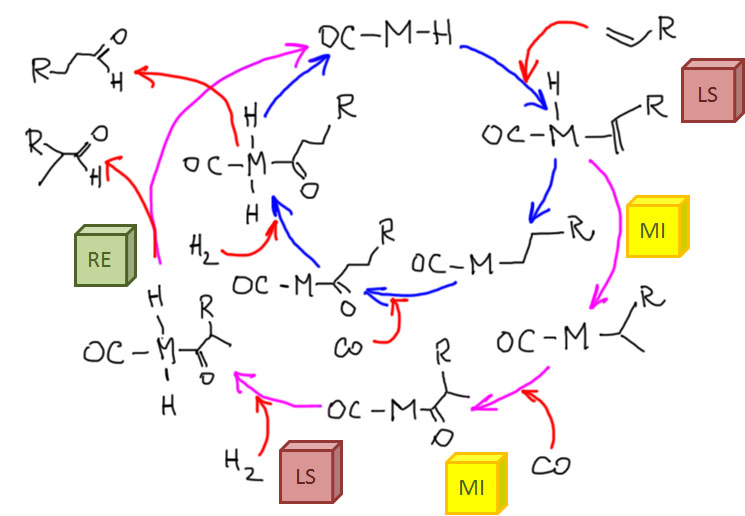
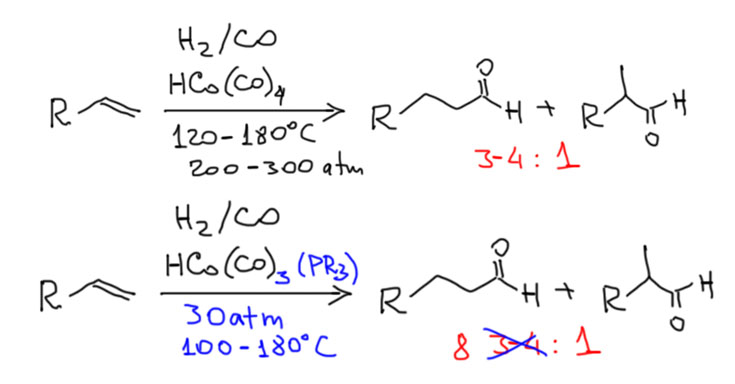
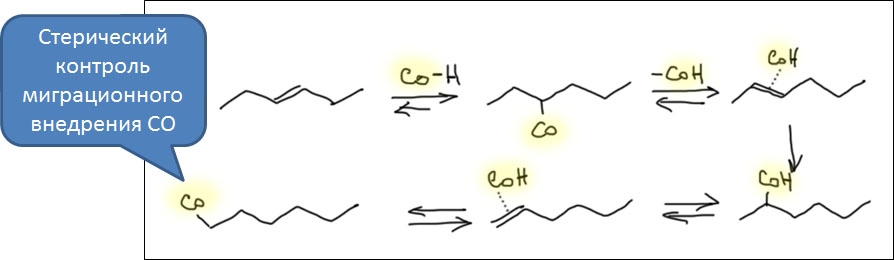
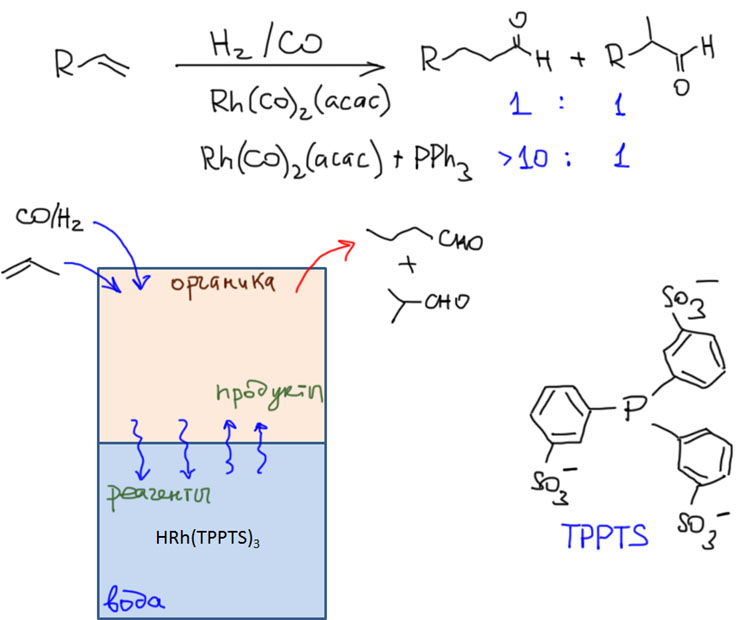
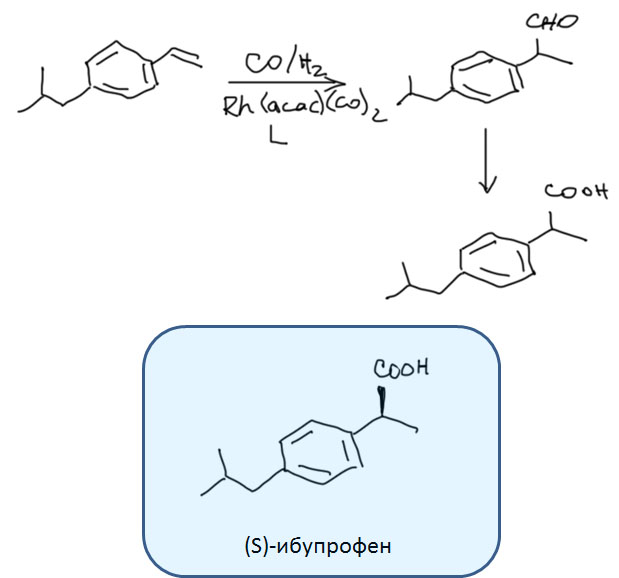
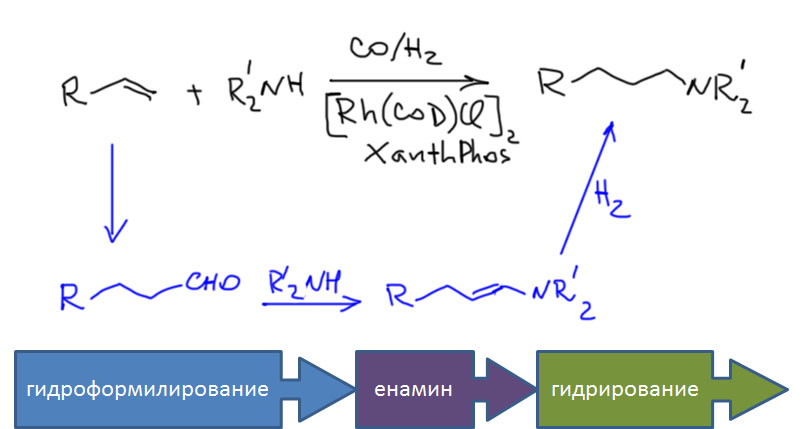
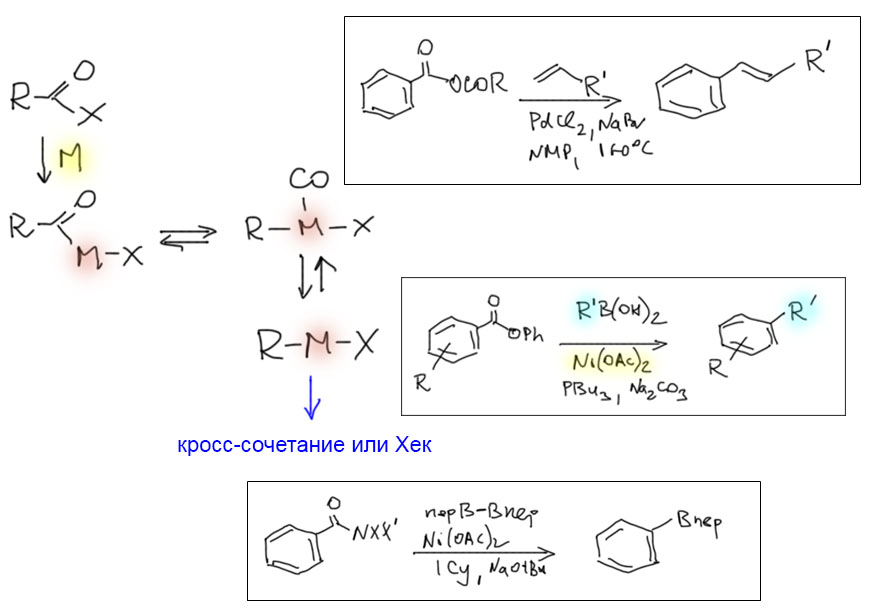
Возможность использовать олефины и ацетилены в реакциях с оксидом углерода открывает прямой путь к превращению важнейшего сырья органического синтеза в альдегиды, кетоны, карбоновые кислоты и их производные. Эту возможность впервые реализовал еще Реппе до Второй мировой войны, и с тех пор работы в этой области не прекращаются. Проблема эта очень сложна, потому что при реакции соединений с кратной связью с участием комплексов переходных металлов возможны многочисленные альтернативные пути реакций с образованием смесей продуктов. Как направить реакцию по одному пути -главная задача разработчика каталитических систем, и здесь требуется очень точное понимание закономерностей влияния структуры координационной сферы. За исключением реакций гидроформилирования, разработка и внедрение которых началось очень давно, все остальное долго висело в состоянии подающего большие надежды, и только в последние 10 лет накопление знаний о механизмах, роли лигандов и всего того, что мы обсуждали все это время, позволило и этой телеге придать неплохой импульс, и она, скрипя и заваливаясь на стороны, наконец поехала.