Уходящие группы в кросс-сочетании и других реакциях
В самом начале химии кросс-сочетания в качестве органических элктрофилов использовали только галогенпроизводные: хлорппроизводные в реакции с магнийорганикой, и в основном бром- и иодпроизводные в остальных реакциях кросс-сочетания. Это сильно ограничивает возможности этого метода. Поэтому с самого начала исследователи приступили к поиску других уходящих групп. Первой был трифлат, и одна эта группа расширила возможности почти бесконечно за счет фенолов и енолизуемых карбонильных соединений. Это было так много, что потребовалось три десятилетия, чтобы это всё переварить. И тогда снова раздался мощный хор голосов: “Ещё давай!!!”. К тому времени началась уже совсем другая химия, новые модные лиганды стали рулить химией переходных металлов, и они позволили начать экспансию в множество уходящих групп: тозилаты, мезилаты и другие сульфонаты, фосфаты, азотные группы и т.д и т.п. Азот из солей диазония долго не давался, потому что имеет дурацкое обыкновение уходить раньше, чем его об этом собираются попросить, но победили и его, хотя это по-прежнему страшно капризная и своенравная штука. Потом пал фторид и уж совсем простые группы типа алкокси. Сейчас ассортимент уходящих групп просто невероятно широк. Соберём понемногу, не торопясь на этой страничке самые важные и интересные. Начнём, естественно, с трифлата.
Трифлаты
Трифлаты – производные трифторметансульфоновой кислоты (по-английски, triflic acid, не имеет перевода на русский). Это одна из самых сильных протонных кислот среди индивидуальных соединений (не смесей типа кислоты Ола), и естественно её анион является одной из лучших уходящих групп. В нашем веке появилось несколько еще более сильных кислот, но их анионы совершенно не годятся как уходящие группы, поэтому монополию трифлата если кто-то и оспаривает, то это его ближайшие аналоги – другие перфторалкансульфонаты, например, нонафлат, но и это в общем-то экзотика для специальных задач. Хорошо и то, что это вполне стабильная группа (в отличие от, например, остатка фторсульфоновой кислоты, сравнимой по силе).
Трифлат обычно не вешают на sp3-гибридный углерод, такие производные не очень стабильны из-за лёгкости сольволиза. Зато на sp2-гибридном углероде это просто находка для синтеза с участием переходных металлов. Такие производные легко получаются и весьма стабильны, вполне выдерживают хроматографию как метод очистки. Введение трифлатов в практику синтеза вполне можно считать одним из выдающихся достижений. Занимался этим в основном Питер Стэнг с сотрудниками в конце 1960х. А в катализ комплексами переходных металлов трифлаты ввёл в 1989 году Джон Стилле прямо накануне свей трагической гибели, и с тех пор они в этой химии совершенно незаменимы.
Трифлаты фенолов получают очень просто действием ангидрида трифторметансульфоновой кислоты на фенолы в присутствии основания, годится пиридин, хотя используют и другие основания, даже карбонаты щелочных металлов. В более современной химии довольно часто используют реагент МакМурри (см. ниже).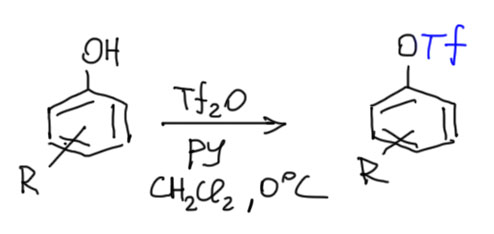
Винил трифлаты – это просто трифторметансульфоновые эфиры енолов. Впервые получил их и предложил как интересный эквивалент винильных катионов в 1969 г Питер Стэнг с сотр. Проще всего их получать по шаблонному методу превращения енолизуемых карбонильных соединений в производные енолов – на равновесный енол действуем хорошим электрофилом в присутствии подходящего основания. Так мы получаем силиловые эфиры енолов, так получем и трифлаты. Используем ангидрид трфторметансульфоновой кислоты.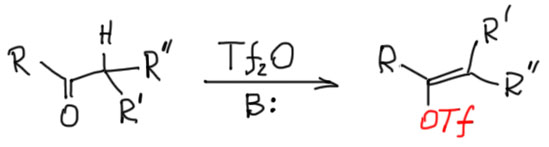
Надо понимать, что ангидрид этой страшной кислоты тоже страшен, уже как ангидрид – его реакционная способность неукротима и чудовищна, работать с ним поэтому нужно очень аккуратно. Реакции с этим агентом начинают при низкой температуре, чтобы при добавлении не произошло перегрева с полным осмолением. Потом медленно отпускают температуру. В качестве оснований можно брать самые простые вещи типа безводного карбоната натрия или пиридина, вот в этом случае он точно должен быть тщательно обезвожен. Если кетон несимметричный, в полном соответствии с равновесной химией енолов (если забыли, можете освежить на моём органическом сайте) получите более замещённый енолтрифлат, к сожалению, всегда с небольшой примесью менее замещённого, но в практических целях этой примесью обычно можно пренебречь.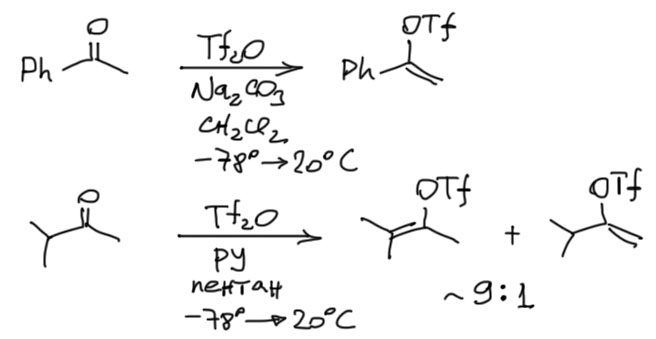
Когда кетоны становятся сложнее, возникает желание сделать реакцию насколько возможно чище. В таких случаях страются подобрать основание, обладающее минимальной нуклеофильностью. В этой химии это очень важно. Тогда в молекулу пиридина вводят заместители в 2- и 6-положения – они отталкивают электрофилы, но не мешают азоту забирать протон – повторю ещё раз, что стерика очень мешает нуклеофильности, но почти не влияет на основность. Самая удачная система этого типа 4-метил-2,6-ди-трет-бутилпиридин (MDTBP), приблизительно на 2 единицы pK более основный, чем пиридин (сказывается индуктивный эффект трёх алкилов), но практически не проявляющий нуклеофильности (даже самый маленький нуклеофил не пролезет мимо двух трет-бутилов до неподелённой пары азота). В присутствии этого основания реакцию можно делать без охлаждения, и получать трифлаты енолов из альдегидов (Stang et al., Synthesis 1980, 283, с другими основаниями они немедленно вступают в самоконденсации и осмоляются) и более сложных соединений, например, ацетиленовых кетонов (Stang et al., Synthesis, 1979, 438). Во втором случае обратите внимание, что трифлат енола не обращает никакого внимания на обработку водным раствором нуклеофила и основания для снятия защиты – трифлатная группа, как уже было сказано, на sp2-гибридном углероде – очень устойчивая штука, не подверженная спонтанному гидролизу. Это очень удобно.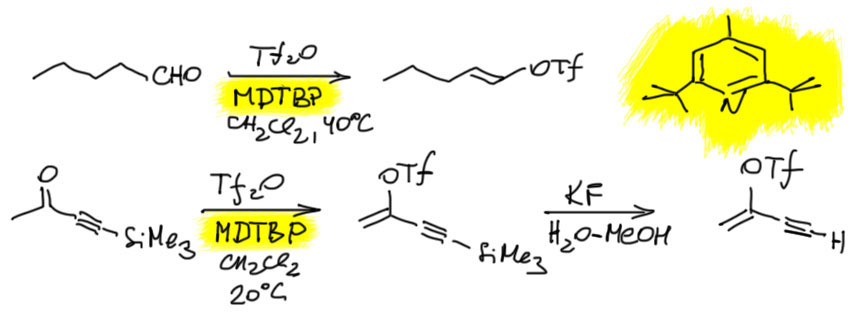
Как и в случае других производных енолов, образование в равновесных условиях не позволяет достичь совсем высокой селективности, а также, в случае несимметричного кетона получить менее устойчивую форму. Тогда мы идём не через енолы, а через еноляты. Способов генерировать еноляты мы знаем много, самый прямой – депротонирование очень сильным стерически-затруднённым основанием типа LDA. Но чтобы превратить енолят в трифлат нужен новый реагент. Ангидрид для этого малопригоден, он слишком крут и при всех предосторожностях, охлаждении и т.п., всё равно получается плохо. Нужен реагент помягче. Такой был придуман МакМурри специально для этой цели, и представляет собой нечто не очень обычное – это анилин, два раза сульфонилированный ангидридом. Ангидрид, как мы выяснили, очень крут, и элементарно это делает в очень мягких условиях. Зачем нужен такой реагент с двумя трифлильными (не трифлатными! трифлат это OTf, а не просто Tf) остатками? Чтобы создать уходящую группу, не самую сильную, но достаточную для того, чтобы мы получили сульфонилирующий реагент умеренной силы. Переносится только одна трифлильная группа, а вторая делокализует минус на азоте (трифлил – один из самых мощных акцепторов, сравнимый с нитро-группой). Реакция идёт мягко при небольшом охлаждении. Реагент МакМурри (McMurry reagent) в этой химии стал стандартом.
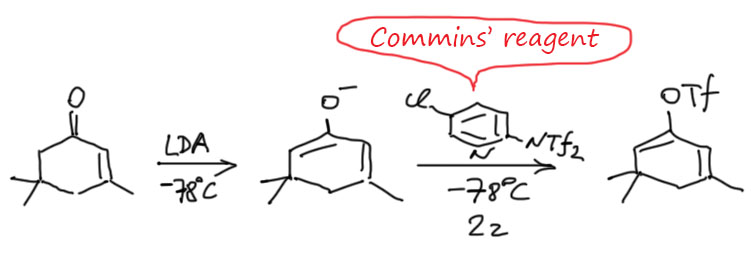
Зачем нужен такой реагент с двумя трифлильными (не трифлатными! трифлат это OTf, а не просто Tf) остатками? Чтобы создать уходящую группу, не самую сильную, но достаточную для того, чтобы мы получили сульфонилирующий реагент умеренной силы. Переносится только одна трифлильная группа, а вторая делокализует минус на азоте (трифлил – один из самых мощных акцепторов, сравнимый с нитро-группой). Реакция идёт мягко при небольшом охлаждении. Реагент МакМурри (McMurry reagent) в этой химии стал стандартом.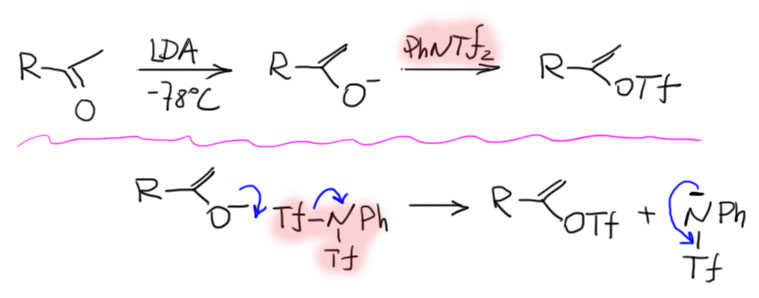
Но нет пределов усовершенствованию. И реагент МакМурри можно сделать лучше. Что в нём не так? Слабоват. Перестарались в ослаблении реакционной способности. Еноляты реагируют долго, несколько часов, и при комнатной температуре. Это удобно, но еноляты не очень любят долго быть при комнатной температуре. Любой малюсенький косяк в эксперименте – немного воды попало через неплотный шлиф, или один из реагентов оказался не безупречен, и из енолята получится немного исходного кетона, и начинается равновесное превращение кинетического енолята в термодинамический, альдольная конденсация и прочие радости. Поэтому реагент нужно немного подкрутить, чтобы немного повысить электрофильность, сделать получше уходящую группу. Самый популярный реагент этого типа называется реагентом Коминса (Comins’ reagent). Это то же самое, что реагент МакМурри, но фенил заменяем на 2-пиридил, в который ещё для дополнительной тонкой подстройки электрофильности добавляем хлор. Скорее всего, дополнительный эффект даёт сближенность трифлильного остатка и пиридинового азота, что должно помогать связывать оба реагента и противоион в такой реагирующий почти внутримолекулярно комплекс. Такие эффекты всегда помогают. Что-то типа этого: 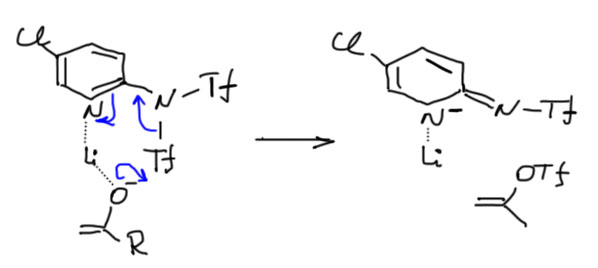 Такая, с виду небольшая настройка даёт удивительный эффект – реакция идёт при -78ºС, то есть как раз при температуре генерации енолята. Выход и селективность повышаются, реагент Коминса завоёвывает практикующих синтетиков. Получим, например, енолтрифлат из непредельного кетона, изофорона.
Такая, с виду небольшая настройка даёт удивительный эффект – реакция идёт при -78ºС, то есть как раз при температуре генерации енолята. Выход и селективность повышаются, реагент Коминса завоёвывает практикующих синтетиков. Получим, например, енолтрифлат из непредельного кетона, изофорона.