
энантиоселективность
DE
Для синтеза очередного природного соединения понадобился в приличных количествах оптически чистый циклопропен, очень маленькая молекула, прямо непривычно маленькая для наших заданий. Но в этом есть немаленькая проблема. Энантиоселективные реакции лучше удаются, когда целью является немаленькая молекула, потому что какой бы ни был конкретный механизм переноса хиральности, в самой сути его всё равно стерические препятствия, разница в стерических препятствиях на двух направлениях атаки на прохиральный элемент. Когда в реакции участвуют маленькие жёсткие молекулы найти серьёзную разницу в стерике непросто и реакций таких очень мало. Здесь использовали энантиоселективное циклопропанирование, которое работает не только с олефинами, но и с алкинами. Метод оказался совершенно великолепен, дал практически количественный выход искомого продукта, причём, что редкость в современной химии, на очень хорошей загрузке не в милиграммах, а сразу аж 15 грамм. И каталитическая эффективность была недурна, загрузка катализатора очень маленькая, всего 0.25 моль%, то есть TON около 400. Здесь это важно, потому что родий дорог, а продукта хотелось побольше, хотя всё остальное в катализаторе весьма бюджетное, но об этом ниже. Реакция шла 6 часов при нуле – охлаждение частс используют в энантиоселективных реакциях, так как это дает дополнительный выигрыш в энантиоселективности – разница в энергиях активации двух конкурирующих путей к энантиомерам редко бывает велика, и снижение температуры приводит к большей разнице в константах скоростей – это элементарное следствие из уравнения Аррениуса. Одновременно вся реакция становится медленнее – и приходится искать компромисс между временем реакции и выигрышем в энантиоселективности. Итак, TOF – 400 циклов за 6 часов – приблизительно 70 циклов в час или одного в минуту. Энантиоселективность тоже неплоха, особенно для такой простой молекулы. Авторы вместо энантиомерного избытка дают энантиомерное соотношение, видимо, для однообразия – дальше у них везде диастереоселективность. Но мы не поленимся, пересчитаем: ee = 94-6 = 88%. Ах, вот оно в чём дело! Авторам не хочется приводить цифры меньше 90% – психологически непросто признать, что недотянули. Такая милая уловочка для того, чтобы в статье мелькали хорошие цифры. 88% ещё и немного смущает, потому что от этого уровня не так просто отчистится, если хочется повысить оптическую чистоту, тем более что продукт – жидкость и его не перекристаллизуешь. Но всё равно всё здорово, метод превосходен, и именно он заслуживает более подробного рассказа, чем мы сейчас и займёмся.
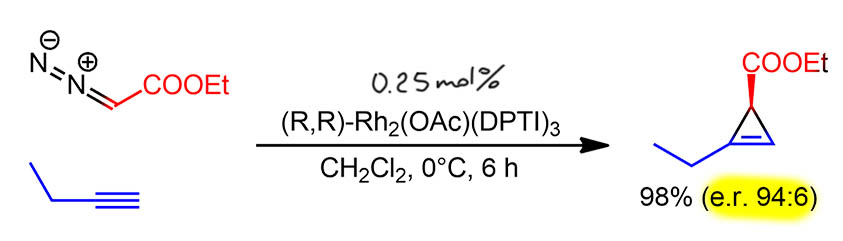
В этой работе успешно использован метод, разработанный выдающимся синтетиком 20 века, Элайей Кори с сотрудниками в 2004 году (Y. Lou, M. Horikawa, R. A. Kloster, N. A. Hawryluk, E. J. Corey, J. Am.Chem. Soc. 2004, 126, 8916). Кори в последний период своей невероятной деятельности увлёкся энантиоселективным синтезом, и сделал в этой области несколько небольших, но чрезвычайно интересных работ, каждый раз предлагая для какой-то очередной большой проблемы частное, но очень яркое и красивое решение. Так и здесь – Кори влез в энантиоселективное циклопропанирование родиевыми карбеноидами. Мы уже разбирали эту область, где катализатором являются комплексы типа “гребное колесо” (paddlewheel), которые легко достигают высокой региоселективности и диастереоселективности, но с гораздо большими трудами – энантиоселективности, хотя несколько интерересных решений было известно, с использованием весьма громоздких хиральных лигандов, но проблем с такими всгда быломного. Кори велик, и ему не нужно мельтешить и доказывать как он велик, городя какое-нибудь чудище – он любил простые вещи и мог себе это позволить. Вот и в этот раз, он предложил ну очень простой лиганд, с хиральностью, заимствованной из продажного реактива, (R,R)-диаминодифенилэтана, из которого просто сделали циклическую мочевину (она же, гетероцикл имидазолидин-2-он, естественно сохранившую два стереоцентра исходного диамина. Дальше нужно было сделать так, чтобы в лиганде осталось только два координационных центра, и был избран самый неожиданный способ – перевод в N-трифторметисульфонильное производной. Почему? Да просто потому, что мочевина это уже амид, нуклеофильность очень низкая, ее очень трудно алкилировать и почти невозможно ацилировать – если не взять чрезвычайно электрофильный ангидрид трифторметансульфоновой кислоты. Поэтому получилось очень просто, но никто бы не рассмотрел в этой немудрёной молекуле высокоэффективный индуктор хиральности.
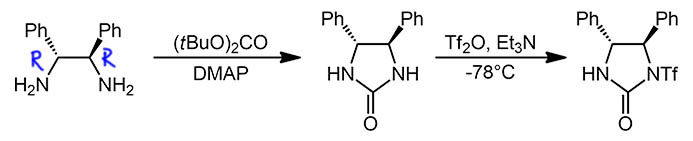
Но он таковым сам по себе и не является. Сначала его надо смонтировать на родиевом кластере. Это очень важно: сам по себе этот немудрёный лиганд никаким эффективным индуктором хиральности не является и являться не может. Перенос хиральности осуществляется только тогда, когда этот лиганд входит в комплекс родия типа “гребное колесо” причём не один, а не менее двух штук – и тогда именно комплекс приобретает хиральность и становится индуктором хиральности.
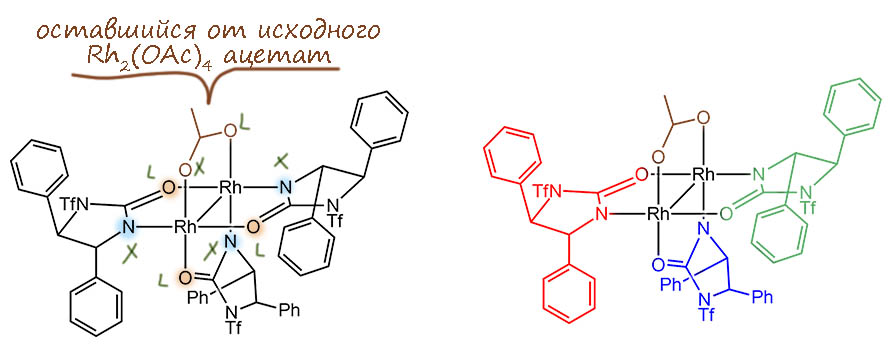
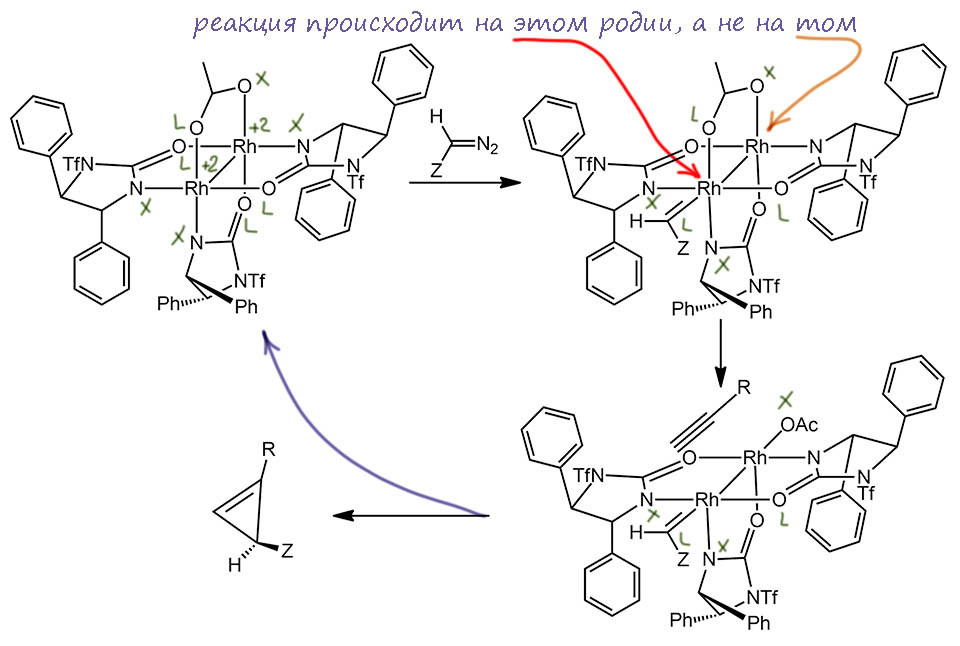
Здесь мы сразу видим, зачем потребовалось делать этот лиганд однобоким – если бы не защита одного азота, каждая молекула лиганда могла бы подойти на диродиевую ось двумя способами, возникла бы смесь диастереомеров, в том числе и симметричных относительно плоскости, проходящей через ось – и это привело бы к потере возможности переноса хиральности даже несмотря на то, что за счёт лиганда внизу комплекс в целом сохранял бы оптическую активность. Вот он, специально нарисованный два раза, первый – чтобы показать, что у каждого родия набор лигандов соответствует типу RhX2L2 и степени окисления +2, оставляющий неспаренный электрон на связь Rh-Rh. Второй рисунок показывает, как однобокий способ связывания хирального лиганда создаёт очень ясное различие между “лево” и “право” вокруг каждого из двух атомов родия. Нижний лиганд, нарисованный синим, практически не влияет на стереодискриминацию возле атомов металла. Можно было бы его заменить вообще на всё что угодно, но это потребовало бы сложного синтеза, и это не то, к чему стремится Кори. Задача должна быть решена просто. Пока это именно так, потому что этот комплекс собрался таким сам. Этот лиганд замещает ацетаты в димерном ацетате родия, но если не форсировать условия, из четырёх лопаток гребного колеса заменяет только три (если поднажать да поднагреть, как любил выражаться мой первый научный руководитель Д.И.Махоньков, то можно заместить все четыре, но Кори делать этого не советует).
И вот это немного недоделанное гребное колесо оказалось совершенно великолепным катализатором энантиоселективного циклопропанирования не только олефинов, но и ацетиленов. Пример такой реакции как раз и есть в обсуждаемом задании. Чтобы объяснить такую выдающуюся работу этой системы, Кори предложил по-новому посмотреть на механизм циклопропанирования. Напомню, что обычно считается, что всё гребное колесо целиком в реакции сохраняется, свободное место на родии цепляет карбеновый лиганд в согласованном процессе экструзии азота из диазоалкана, и дальше такой карбеноид реагирует с олефином или ацетиленом тоже в согласованном процессе, когда сам металл с кратной связью прямо не взаимодействует. Такой механизм неплохо объясняет региоселективность и диастереоселективность, но не очень хорошо – энантиоселективность, так как само взаимодействие карбеноида и кратной связи происходит не очень близко к тем областям катализатора, где сильно нарушается симметрия. Кори предположил для своего катализатора, что сначла происходит расщепление хелатного мостика у ацетата – мы бы назвали это проявлением гемилабильности, но в 2004 году это слово ещё не вошло в моду. Дехелатирование ацетата освобождает ещё одно место на одном из атомов родия, и на это место садится олефин или ацетилен. И с учетом несимметричного окружения этого атома и карбен и субстрат размещаются однозначно – чтобы минимизировать стерические препятствия. Вот как это нарисовано в работе, немного подробнее, но с сохранением взаимного расположения участников событий:
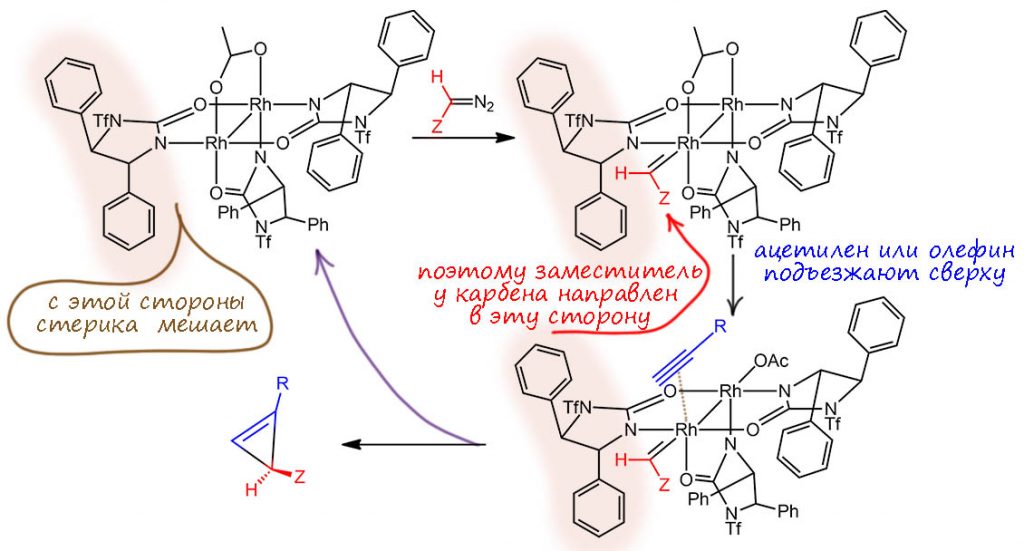
Видим, что родиевый комплекс, как обычно, вызывает расщепление диазосоединения и цепляет карбен. При этом, несимметричная форма комплекса такова, что с одной стороны лиганд развернут вперёд, а с другой – назад, и тогда заместитель на карбене вынужден располагаться совершенно определённым образом. После этого и происходит расхелатирование мостикового ацетата, видимо, потому что связывание карбена ослабляет координационные способности металла. На свободное место приходит олефин или ацетилен, и опять форма комплекса направляет его определенным способом, чтобы уменьшить стерические взаимодействия заместителя на олефине/алкине – продукт поэтому получается с вполне определенной конфигурацией. Собственно перенос хиральности происходит при взаимодействии ненасыщенного субстрата с карбеном только с одной прохиральной стороны – возникает стереогенный центр.
Красиво? Красиво! Но тут глаз падает на один из атомов родия и возникает какой-то смутный дискомфорт. Смотрим на схему внимательно. Ой, а почему расщепление хелатного цикла происходит с переднего атома, а не с заднего? Ой, а какова степень окисления переднего родия? Ведь ацетатный мостик именно сюда цеплялся X-способом, тогда здесь будут два X и два L-лиганда, а после связывания карбена – три L-лиганда. Если так, то после расщепления мостика на этом родии должен возникнуть плюс. А на заднем – а там всё наоборот, там уже было два X-лиганда, и если придет еще ацетат, то будет минус (степень окисления родия измениться при расщеплении хелата не может!). Тут явно что-то не то. Конечно, можно расставить заряды, ничего страшного в этом нет, ведь они всё равно формальны, а в реальной молекуле как-то затейливо распределятся по атомам вокруг. Но как-то это неуклюже.
Давайте покритикуем Кори. Приятно же, чёрт возьми! Вспомним, как в советской школе, к которой мы нынче так мечтаем вернуться, дети малые смело критиковали Толстого и всех остальных, кроме Ленина и текущего генсека, естественно, – и сочинения писали: “Толстой не понял … значение диктатуры пролетариата в социалистической революции”, “Пушкин не сумел предвидеть руководящую роль партии” “Критика капитализма в пьесах Чехова лишена глубины анализа, данного в отчетном докладе генерального секретаря имярек съезду партии номер такой-то” и т.п. Так что с таким-то опытом за плечами нам ли какого-то Кори стесняться, хотелось написать, англо-саксонского, но как на беду, Кори – чистокровный ливанский араб урождённый Аль-Кхури! Вот и мы посмотрим на картинку, нарисованную в замечательной статье, и увидим, что Кори не помешало бы послушать лекцию по координационной химии. Или хотя бы кому-то из его соавторов. При этом мы полностью согласимся с тем, что мысль этой работы очень хороша, и чуть ниже перерисуем картинку правильно и немного додумаем за авторами их замысел. При этом не будем выпендриваться уж слишком сильно, не будем забывать, что Кори в момент написания той статьи было 76 лет, и можно только мечтать сохранить хотя бы небольшую часть творческих способностей в таком возрасте. К тому же великий патриарх и в 2022 году ещё не присоединился к сонму богов органической химии, и пока еще остается среди смертных самим своим присутствием намекая некоторым на то что большой хирш ещё не гарантия бессмертия (у Кори хирш колоссальный, но это не главное).
На самом деле, тут все очень легко переделать так, чтобы и законы координационной химии не пострадали и механизм не пропал, а даже наоборот засиял новыми красками. Ведь в нём заложена ещё одна очень интересная фишка, которую Кори, кажется, не заметил.
Фокус в том, что в таком комплексе, который получили Кори с сотрудниками, атомы родия сразу разные. Структуру комплекса они установили по рентгену, но как он дальше работает – это просто правдоподобная гипотеза. Поэтому мы не обязаны повторять схему из статьи, но при этом должны использовать доказанную структуру. Когда эти гребные колёса делают из карбоксилатов, карбоксилаты – симметричные хелаторы, и мы произвольно назначаем кислородам X и L-функции, чтобы получить симметричную структуру. Но здесь лиганд заведомо, изначально несимметричен, кислород у него L-лиганд, а азот – X-лиганд, азотный центр заведомо более донорный, чем кислородный, и наводит на металл большую электронную плотность. А поскольку таких лигандов у нас нечётное количество, мы не можем распределить их поровну – тебе половинку азота, и тебе половинку. У одного родия два азота, и это уже исчерпывает степень окисления. У другого – один. Кори нарисовал структуру так, что впереди оказался тот, у кого один, насколько можно судить, совершенно случайно – и с этим и возникли проблемы. Но если подумать, логичнее был бы другой выбор. Хотя бы потому что ключом к механизму Кори является гемилабильность ацетата – но гораздо логичнее ожидать расщепления хелатного цикла со стороны атома металла, окруженного более дорными лигандами, то есть того, на котором два азота, а не один. Чтобы так поступить, удобно развернуть комплекс на 180 грудусов, и тогда мы увидим, например, что симметрия окружения атомов не изменилась – это логичное следствие гомохиральности, делающей молекулы напоминающими спираль, а спираль как ни верти, левая останется левой, а правая – правой. И можно даже не крутить, а просто перевернуть нижний лиганд – он намсам по себе до лампочки, и переворот его ничего не изменяет – получается тот же комплекс – здесь нет никакой новой стереоизомерии. Когда мы это сделаем, мы спокойно сотанемся с тем же механизмом, только уже без вопросов – а где заряды, а почему хелат раскрывается так странно, что возникают заряды. И даже более того – мы поймём, что именно такая структура комплекса и была причиной возникновения возможности гемилабильности ацетата – более донорный атом родия имеет меньшую константу связывания с атомом кислорода, и когда на металл садится ещё и карбен, тоже донорный лиганд, такое расхелатирование становится вполне разумным продолжением реакции. Всё остальное мы уже рассмотрели на первой схеме механизма.
диастереоспецифичность
DV
Энантиоселективное гидрирование на родиевом катализаторе. С одной стороны, это весьма типичный кейс энантиоселективного гидрирования, только некоторыми нюансами отличающийся от классики Ноулза-Ноёри, получившей в 2001 году нобелевку. С другой стороны, это удачный случай еще раз разобрать, что такое энантиоселективность и диастереоселективность, и разобраться в несколько парадоксальном результате – в этой работе получен именно диастереомер, но реакцию не квалифицируют как диастереоселективную, а называют только энантиоселективной.
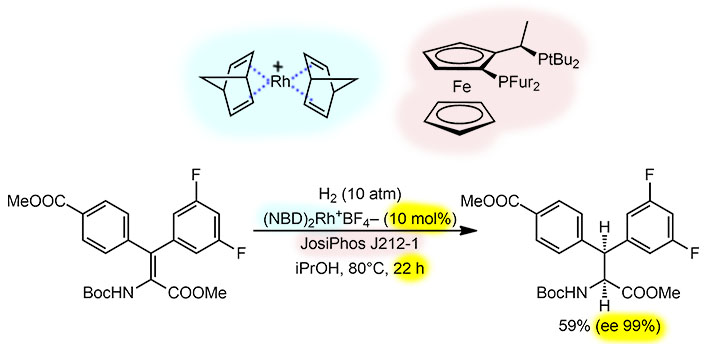
Цель – получить оптически чистые аминокислоты, содержащие два разных ароматических заместителя на α-углероде. Мы просто явно узнаём ту задачу, которая стояла перед основоположником этой химии Ноулзом, когда он разрабатывал процесс для промышленного производста до сих пор основного лекарства, отсрочивающего развитие симптомов болезни Паркинсона, леводофы (левовращающего дигидроксифенилаланина). И сам этот подход – энантиоселективное гидрирование дегидроаминокислот, защищенных по амино и карбокси-группам с тех пор стал основным и таким способом получены тысячи искусственных аминокислот, весьма популярных строительных блоков для разработки новых лекарственных препаратов. Так что же особенного в этой работе и почему она появилась совсем недавно, ведь методу этому уже сильно больше сорока лет. Проблема в двух арилах, в том, что они создают второй стереогенный центр, а следовательно должны в исходном ненасыщенном предшественнике быть стереохимически определёнными. Эту задачу в этой работе успешно решили с помощью кросс-сочетания Судзуки-Мияуры, и это позволило наработать есколько десятков определенных E/Z-диастереомеров. Второе – ктализ на переходных металлах не любит избыточную стерику и гидрировать тетразамещённую двойную связь сильно непросто. Даже здесь мы видим необычно жесткие для гидрирования условия – давление водорода, хоть и небольшое, и повышенную температуру. Олефины попроще гидрируются намного легче. А повышенная температура, которая здесь, видимо, необходима, чтобы реакция шла с разумной скоростью (даже в заявленных условиях реакция занимает 22 часа!), наклабывает дополнительные требования на энантиоселективный катализатор – он должен быть особенно хорошо стереодифференцирующим, различающим по энергии два подхода к прохиральным сторонам плоской молекулы, иначе повышенная температура просто нивелирует разницу в энергиях активации, которая в энантиодифференцирующих процессах никогда не бывает слишком велика. Понятно, что задача чертовски непроста – и стерику одолеть, и энантиодискриминации (специально использую кучу синонимов, которые используются в асимметрическом катализе) хорошей, а лучше отличной, добиться. Одна задача требует не слишком нагружать катализатор объёмистыми лигандами, другая – наоборот зовёт хиральные лиганды, обеспечивающие сильно разную стерику с двух сторон подхода. Как искать такие лиганды и вообще каталитические системы, ведь и форма предкатализатора тоже очень важна. Увы, никакой теории на этот счёт мы так пока и не имеем, расчеты тоже помогают мало. Поэтому в ход идет то, чем реально обогатилась химия в новом веке – возможностями просто ставить и обрабатывать сотни и тысячи сравнительных экспериментов – автоматизированный высокопроизводительный (high throughput) скрининг.Коммерческие компании предлагают готовые наборы сотен лигандов, и все их проверяют на микрозагрузках – только чтобы пустить реакционную смесь в хромато-масс-спектроскопию. И такой подход дает результаты – находятся эффективные каталитические системы и лиганды, тогда уже их исследуют получше на разумных загрузках.
Здесь использовали в качестве предкатализатора катионный комплекс родия с двумя норборненами – типичный для d8-конфигурации металлов 9-й и 10-й групп плоскоквадратный комплекс; норборнены в условиях гидрирования моментально прогидрируются сами и оставят родий наедине с добавленным лигандом, который хотя и бидентатен, но зело рогат и две штуки в координационную сферу палладия не лезут, лезет один, и родий остается координационно-ненасыщенным, весьма электрофильным – и это помогает справляться с громоздкими олефинами для гидрирования. Так дело пошло и было найдено сразу несколько отличных лигандов, все на платформе ферроцена, и дале среди них выбрали те, что подешевле и есть в продаже. Такими окзались лиганды семейства Жозифос, мы уже разбирались, откуда они взялись и почему они, несмотря на грозный вид довольно доступны. Целые наборы лигандов этого семейства продает одна швейцарская компания, они отличаются заместителями в обоих фосфинах: в данном случае лучше всех себя показал тот, у кого 2-фурилы в одном фосфине и трет-бутилы – в другом; этот лиганд по устоявшейся традиции назвали бы FurPF-tBu, но у всех Жозифосов есть цифровой код, с которым их и продают. Оценим результат: загрузка предкатализатора немаленькая, 10 моль%, поэтому TON совсем невелик, всего 6 (шесть, вспомним как жадно сосал водород классический катализатор Уилкинсона, – “не успеваем грушу наполнять”, – жаловался великий учёный, но там олефины были попроще). И это за 22 часа, то есть почти по 4 часа на цикл, TOF – четверть цикла в час. Но оптическая чистота великолепна, и только скромность, видимо, не позволила написать 100 процентов.
Ну и теперь самое интересное – это энантио или диастереоселективность?
Ответ вроде бы очевиден – и то, и другое. Но здесь есть довольно важный нюанс. Селективность – это от слова выбор, когда есть два или больше альтернативных путей реакций, не очень сильно отличающихся по энергиям активации, и поэтому получается смесь продуктов, и тогда идет игра на повышение выхода одного из продуктов за счет другого или других. И тогда мы говорим о селективности, энантио- или дистерео- или просто стерео-, или другой. Но бывают реакции, в которых результат предопределён. Вот, например, в SN2-замещении обращение происходит свегда, хоть тресни, и на 100%. Не бывает такого, что на 95% обращение, а на пять сохранение. Вернее, бывает, но мы тогда делаем вывод, что механизм реакции не SN2. Или что есть два конкурирующих пути, главный SN2, но есть и другой, например, перенос электрона, что-то типа SRN1. Но если путь один и это SN2, то нет смысла говорить о стереоселективности – в таких случаях говорят о стереоспецифичности: реакция сопровождается стереоспецифическим обращением конфигурации. Вот этот термин “специфичность” иногда понимают так, что это очень большая, почти стопроцентная селективность. Но это неправильно и не соответствует смыслу этого термина: настоящий смысл состоит в том, что течение реакции определено природой механизма, то есть у реакции не два или более конкурирующих путей, а только один, по которому она и следует.
Итак, специфичность это отсутствие селективности по причине единственности пути реакции. Выбора нет, или он такой как в некоторых политических системах – вот вам царь-государь, извольте выбрать и не выпендривайтесь, а то…! Интересно, что в обычной органической химии мы не так много видели таких механизмов. Замещение SN2 – это да, это классика. А что ещё? В элиминировании и присоединении все эти син и анти как правило это именно селективность, преобладающий, но не единственный путь. Да, в согласованных реакциях, например, гидроборировании путь тоже предопределён, и специфичен. Получается, что специфичность часто работает именно в сограсласованных механизмах. Но в химии переходных металлов, как мы уже видели, таковых большинство. Поэтомув химии переходных металлов мы очень часто видим именно специфичность. Но какую? Вот гидрирование на родиевом катализаторе. Мы знаем, что гидрирование на родии идет по дигидридному механизму – сначала водород садится двумя гидридами на родий, затем упрощённо мы видим такой механизм – гидрородирование двойной связи, за которым следует восстановительное элиминирование первая реакция идет стереоспецифично син, вторая стереоспецифично с сохранением конфигурации: результат – стереоспецифично син.
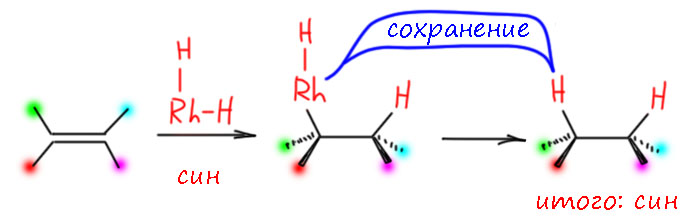
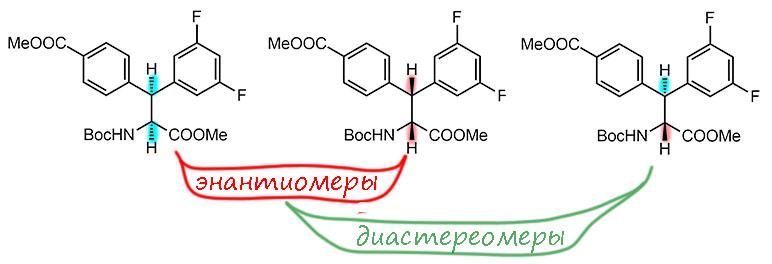
В применении к рассматриваемой реакции мы видим, что оба атома водорода садятся по одну сторону от плоскости бывшего олефина. Выбор этой стороны – это энантиодифференциация, результат – энантиоселективность. Но то, что оба водорода по одну сторону не подвергается сомнению: диастереомер получается один, энантиомерный состав этого диастереомера модет быть разным, но он сам один. Эта реакция диастереоспецифична и энантиоселективна. Поэтому мы не указываем показатели диастереоселективности, в виде отношения диастереомеров – указывать нечего, он один, сто – ноль. Энантиомеры получаются, и с другими лигандами энантиоселективность не так велика, образуются оба. А вот диастереомер не получается вообще, даже с самым энантио-неселектиным лигандом. Вот какая ситуация с диастереоспецифичностью.
Обязательно ли родиевое гидрирование диастереоспецифично? Нет, во-первых, мы не застрахованы от вмешательства другого механизма, какого-то, мы априори не можем этого отрицать. Во-вторых, и это реально происходило не раз, олефин может изомеризоваться в условиях реакции, например, под действием какой-то другой формы комплекса, скажем, просто кислоты Льюиса. И после изомеризации само гидрирование может сколько угодно быть диастереоспецифичным, но реакция в целом перестанет быть таковой, просто потому что это уже не одна реакция, а две.
Вообще со стереохимией надо работать аккуратно и не путать конфигурации. В той же работе есть второй диастереомер, полученный из другого диастереомера олефина – с ним получился побольше выход, 71%, но меньше энантиоселективность, ee 92%. Путать нельзя – химия не терпит неряшливости в стереоконфигурациях.
энантио/диастереоселективность
DA
Очень интересный кейс, отлично показывающий, что энантиоселективность может возникнуть из самой простой реакции без участия хиральных реагентов, и очень тесную связь между диастереоселективностью и энантиоселективностью. В общем виде эта связь подробнее разобрана ниже, но здесь отличный пример. Итак, есть не очень длинный синтез природного соединения, эупоматилона, это такой метаболит одного растения, относящийся к классу лигнанов. Растения для прочности умеют делать очень интересный сильно сшитый полимер лигнин, из которого строится древесина. И пока они этот лигнин конструируют, некоторые кусочки идут в дело, чтобы сделать еще очень полезные небольшие вещества, у которых куча разных функций, от защиты от врагов до инструментов тонкой регулировки жизнедеятельности. У каждого уважающего себя растения, а растения все себя очень уважают, есть свой набор таких лигнанов, так что для исследователей это совершенно бесконечная кладовая – выделяй, изучай, синтезируй не хочу.
Это лигнан сделали довольно быстро, молекула не очень сложная. По дороге пригодилось и кросс-сочетание, но последнюю точку удалось поставить самым простым гидрированием по Уилкинсону в самом исходном его варианте вообще без затей. И это последняя стадия синтеза – продукт этой стадии и есть цель синтеза, в оптически чистом виде, с конфигурацией, соответствующей природному соединению. Сначала прикинем параметры катализа. Загрузка пред-катализатора велика, 30 моль%, и при высоком выходе в 90% это всего три цикла. Нетрудно представить, что в этом синтезе задачи экономить катализатор просто не было, тем более, что загрузки были небольшими. TOF тоже удобно получился – на 3 цикла 3 часа, цикл в час. Реакция не очень быстрая и это понятно – олефин гем-дизамещённый, и с одной стороны довольно объёмистый. Собственно, эта объёмистость очень нужна, потому что именно она обеспечивает диастереоселктивность – гидрирующий катализатор, а это немаленький комплекс родия подходит с обратной стороны от той, куда отогнуты и метил на соседнем атоме и ароматическое кольцо со всем обвесом на следующем. Такая дискриминация оказывается очень эффективной – диастереомер с менее замещенной стороны получается в отношении 20:1, то есть приблизительно 95:5. Это и есть мера диастереоселективности. Она хороша. Бывают больше, и ясно, что если бы взяли родиевый катализатор пообъёмистей, а с такими мы уже здесь встречались и вообще их пропасть, то можно было бы шутя вырулить на 99:1. Подозреваю, что это просто нафиг никому не нужно было. Разумная достаточность – отличный принцип для осмысленной работы в химии.
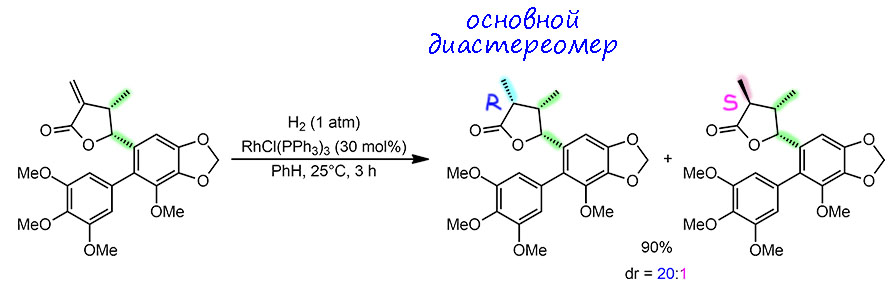
Теперь зададимся вопросом – а это энантиоселективная реакция? Кажется, что нет, ведь энантиомер продукта в этой реакции получиться просто не мог – конфигурации двух других стереоцентров заданы и не изменялись. Хорошо, но новый стереоцентр получился в определенной конфигурации (здесь R), и продукт, у которого этот стереоцентр в противоположной конфигурации (S) образовался в существенно меньшем количестве. Это прямо означает, что произошёл перенос хиральности, не от катализатора, а от уже существующих стереоцентров в молекуле: продукт оптически активен, его оптическая активность определяется конфигурацией трёх стереогенных центров. Два из них остались в том же сотсоянии, что были, но третий получил преимущественно одну конфигурацию. Это ровно она и есть – энантиоселектвность, но проблема в том, что поскольку мерой этой селективности является не соотношение энантиомеров, а соотношение диастереомеров, ее нельзя определить по углу вращения продукта, посколькуу диастереомеров удельные углы совсем разные и никак не связаны простым соотношением плюс-минус. Тем не менее, для единообразия работы с двумя типами стереоселективности, мы имеем право определит энантиомерный избыток точно так же как мы это делали в случае, когда в реакции возникала пара энантиомеров – как разницу между продуктом с одной конфигурацией нового стереоцентра и продуктом с противоположной конфигурацией: ee = 95% – 5% = 90%. Видим, что энантиоселективность неплохая, но не фонтан. Ну, мы другой и не ждали в ситуации, когда энантиодискриминацией мы были обязаны диастереодискриминации от уже имеющихся стереоцентров, ближайший из которых это скромный метил. Вот такая энантиоселективность без энантиомеров.
DO
Интересный пример энантиоселективного и диастереоселективного аллильного замещения. Здесь мы можем очень хорошо видеть, как работают эти два типа стереоселективности, и что нужно сделать, чтобы их не путать. Но кроме этого здесь есть и еще одна фишка. Это не совсем классическая реакция Цудзи-Троста, в которой нуклеофил извне атакует аллильный комплекс. В данном случае нуклеофилом формально является гидрид, но гидриды извне не приходят (я на днях напишу об этом на сайте orgchem.avchem.ru в разделе Вопросы и ответы – так до сих пор и не написал, но обязательно напишу), и здесь он сначала попадает в координационную сферу палладия как лиганд, и далее внутримолекулярно пересаживается на углерод. Как гидрид попадает на металл? Это давно и хорошо известная реакция, которая активно используется в так называемом transfer hydrogenation (попробуйте сами перевести это на русский так чтобы было не очень коряво хотя бы, я не могу) – это когда водород для гидрирования берется не из молекулярного водорода, а из каких-то молекул-переносчиков, с которых водород сначала переезжает на металл, образуя гидридные комплексы, а затем и на субстрат. Этим способом можно избежать использования газообразного водорода в лаборатории, а это, поверьте, та ещё морока, если в вашей лаборатории нет уже готовой разводки этого газа. В качестве переносчиков водорода используют десятки разных соединений, но одни из самых популярных, такая классика этой химии – муравьиная кислота в присутствии оснований, то есть формиат. Мы сейчас не будем разбирать тонкости этой реакции, которая часто встречается у разных переходных металлов, но особенно свойственна поздним – палладию, рутению и т.п. Важно то, что формиат как лиганд в равновесной реакции превращается в гидридный комплекс и диоксид углерода. Эта реакция связана с многими важными процессами, в том числе реакцией водяного газа, восстановления диоксида углерода и т.п. Очевидно, что это очередное ретро-миграционное внедрение. Впрочем, с тем же успехом это можно рассматривать как внутримолекулярную электрофильную активацию C-H связи. Металл не изменяет степени окисления.
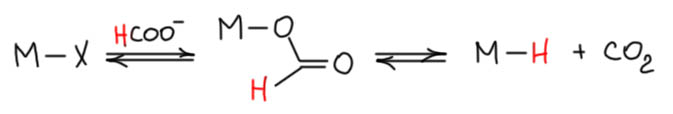
Дальше эти гидридные комплексы участвуют в реакциях гидрирования или гидрогенолиза самых разных субстратов, олефинов, карбонильных соединений, бензильных производных и т.п. В нашем случае это – по типу реакции гидрогенолиз связи C-O, со смещением двойной связи, аллильной перегруппировкой, в реакции формально являющейся нуклеофильным замещением с донором гидрида – SN2′. Почему в одних реакциях двойная связь смещается, в других нет, а в третьих оба направления конкурируют – это в каждом случае решается по-своему, где-то работает стерика, где-то электронные факторы, где-то кинетика. Это довольно мутная химия, и в каждой конкретной разновидности этой реакции есть свои соображения, но часто и просто обобщение наблюдений – получается, и хорошо, а почему знают только всемогущие боги.
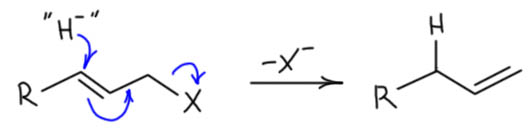
Диастереоселективность и энантиоселективность
Если в результате смещения двойной связи возникает третичный атом углерода, возможна энантио- и/или диастереоселективность, и появляются возможности для переноса хиральности. Посмотрим подробнее. Вот субстрат – атом углерода, на который нацелился гидрид, тригональный, sp2-гибридный, плоский. Но если гидрид на него сядет, он станет тетраэдром, и если два заместителя различны, то и стереогенным, или как мы часто говорим, хиральным (почему термин “стереогенный” корректнее термина “хиральный” мы разбираем в стереохимии). Поэтому в исходной молекуле этот центр называется прохиральным, и мы видим два направления атаки гидрида на этот центр.
- Если в реакции нет ничего хирального, то оба направления по определению и по законам природы равновероятны – в результате получается равные количества двух продуктов. Эти продукты являются энантиомерами, и получается рацемическая смесь.
- Если в реакции есть что-то хиральное, скорее всего, в катализаторе, но может быть и в каких-то других реагентах, участвующих в процессе, но не в самом субстрате, то два направления реакции могут перестать быть равновероятными; для этого безусловно нужно, чтобы хиральная молекула участвовала в механизме реакции, входила в состав переходного состояния – тогда мы получим смесь с преобладанием одного из энантиомеров, и будем характеризовать ее, как мы умеем, энантиомерной или оптической чистотой – просто разностью содержания в процентах большего и меньшего энантиомера.
- Если что-то хиральное есть в субстрате – еще один или несколько стереогенных центров (возможно, и других элементов – осей или плоскостей, но это всё же экзотика, хотя уже давно и нередкая) – то продукты будут уже не энантиомерами, а диастереомерами, и этот случай чрезвычайно распространен в стереоселективном синтезе, и представляет некоторые сложности в анализе. Данная задача ровно про это.
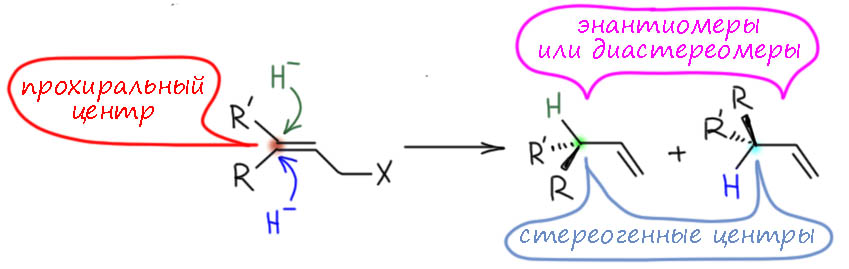
Займёмся диастереселективностью. Вначале на упрощённой модели, а в конце применим знания к задаче DO. Упростим субстрат до минимального набора заместителей, достаточного для анализа; один заместитель заменим на метил, а в другой введём простой стереоцентр. И сразу увидим возможность двух ситуаций. Сначала посмотрим на реакции, в которых нет другого источника хиральности кроме того, который есть в субстрате на уже имеющемся стереоцентре.
Реакции без участия хиральных реагентов (кроме субстрата)
Стереоцентр определён – в реакцию ввели чистый энантиомер субстрата. В реакции конфигурация этого центра не изменяется. Эта оговорка нелишняя, потому что так бывает не всегда, и частичная потеря конфигурации, рацемизация нередко встречается, ведь в реакции могут происходить и всякие непредусмотренные вещи – кислоты и основания могут вызывать обратимые реакции переноса протона, енолизаций и т.п., а переходный металл может вызывать миграцию двойных связей, тоже обратимую. Делаем реакцию. Прохиральный центр (он продолжает называться именно так, несмотря на то, что молекула в целом хиральна из-за стереогенного центра на соседнем атоме, но сам реакционный центр не является стереогенным, но превратится в ткой только после присоединения гидрида и ухода двойной связи) превращается во второй стереогенный, теперь у нас возникает два диастереомера, можем назвать их эритро и трео – кстати, лишний раз убедимся, как удобна эта система, для неё не надо знать старшинства и она вполне годится для общих рассуждений, когда мы вообще ещё не знаем природу заместителей. Первый вопрос – диастереомеры поучатся в одинаковых количествах? Этот вопрос касается диастереоселективности, выражаемой отношением диастереомеров, в любом порядке, например, эритро : трео. Если это соотношение не равно 1:1, реакция диастереоселективна. Как правило, диастереоселективность наблюдается, не обязательно большая, но хотя бы какая-то. Причина этого очевидна – два подхода к прохиральному центру более не равновероятны, потому что отличаются стерические взаимодействия – геометрия стереоцентра рядом несимметричная. По связи, соединяющем этот стереоцентр с реакционным центром, есть конформации, и, как мы знаем из геометрии молекул с внутренним вращением, одна из этих конформаций всегда более выгодна чем остальные, и реакцию можно считать происходящей именно с таким конфомером.
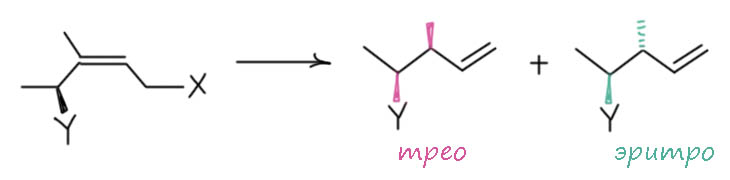
А как насчет энантиоселективности? Конечно, если реакция диастереоселективна, а имевшийся стереоцентр определен, то возникающий стереоцентр будет возникать преимущественно в одной из конфигураций – и это по определению она самая, энантиоселективность. Поскольку у нас продукта только два, два диастереомера, которые мы назвали для простоты трео и эритро, то у каждого из них новый стереоцентр будет иметь конфигурацию R и S. И с точки зрения энантиоселективности, нас интересуют именно они. Разница в том, что диастереоселективность мы характеризуем соотношением диастереомеров, то энантиоселективность мы характеризуем энантиомерным избытком. Давайте пример придумаем. Во-первых, представим, что на последней схеме заместитель Y задаёт старшинство (хлор или гидроксил, например). И в какой-то реакции у нас получилось 90% трео и 10% эритро. Диастереоселективность тогда 90:10 = 9:1. В трео-форме в нашем примере новый стреоцентр имеет конфигурацию (S), в эритро-форме – (R). Энантиоселективность характеризуем энантиомерным избытком, который равен просто разности: ee = 90 – 10 = 80%. Как видим, диастереоселективность нам льстит больше энантиоселективности. Понятно, что дело просто в принятой методике характеристики этих типов селективности. Но одна вещь здесь важна. Когда мы имеем дело с чистой энантиоселективностью, образуются энатиомеры, то энантиомерный избыток является характеристикой оптической чистоты продукта, измеряемой удельным углом вращения – эти две величины связаны очень просто, умножаем ee на удельное вращение чистого энантиомера с тем же знаком, что у продукта – получаем удельное вращение продукта, ну и наоборот – делим удельное вращение продукта на удельное вращение чистого энантиомера с тем же знаком, получаем энантиомерную чистоту (не забываем переводить доли в проценты и наоборот). Если же энантиомерный избыток определяется в реакции, дающей диастереомеры, то это перестает работать, потому что у диастереомеров разное вращение, оно вполне даже может иметь один знак. Если мы знаем удельные вращения обоих диастереомеров, то понятно, что мы справимся с арифметикой и сможем пересчитать угол вращения продукта и в диастереоселективность и в энантиомерный избыток, но это будет хоть немного, но сложнее, и потребует знания не одной величины, а двух, потому что из удельного вращения одного диастереомера невозможно определить вращение другого – его можно только измерить, выделив и очистив оба. Конечно, в наше время величины сооотношения диастереомеров определяют не по углу, а по непосредственному измерению содержания обоих диастереомеров в смеси или по ЯМР, или из хроматографии.
Второй очень распространенный кейс. В реакцию ввели рацемат субстрата. Реакция та же, что в предыдущем кейсе. Понятно, что сама природа хиральности говорит нам, что энантиомеры реагируют с нехиральным реагентом с одинаковой скоростью, образуя продукты симметрично друг другу, так, что раз в реакцию вступил рацемат, то и продуктом будет рацемат. Но диастереоселективность никуда не делась. Разрисуем:
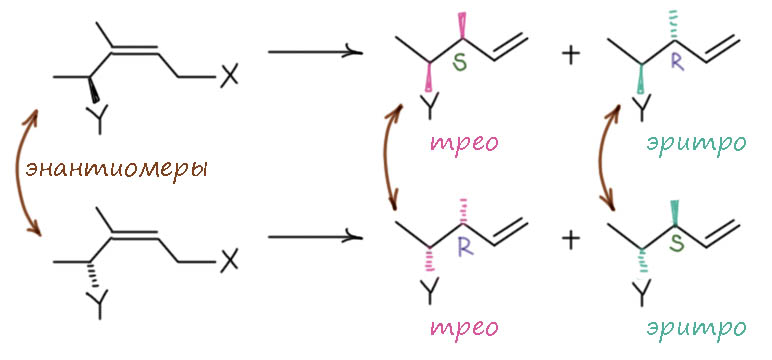
Когда в реакции участвует рацемическая смесь субстрата, реально идут две параллельные реакции с каждым энантиомером. И если в смеси нет других источников хиральности, симметрия не может нарушаться – обе реакции зеркально симметричны – соответствующие продукты образуются так, чтобы не нарушалось отсутвтие оптической активности. Но у каждого энантиомера диастереомерные продукты образуются в разных количествах – мы уже сделали ту же реакцию для чистого энантиомера и понимаем, что второй энантиомер даст то же самое, только зеркально отраженное. Повторим пример, придуманный нами для чистого энантиомера: получается 90% трео и 10% эритро. Второй энантиомер даст то же самое – 90% трео и 10% эритро, только энатиомерных. Итого из всей рацемической смеси получаем 90% трео-рацемата и 10% эритро-рацемата. Диастереоселективность 90:10 = 9:1. Никуда не делась! При том, что никакой оптической активности не возникло и не могло возникнуть, а для каждого из энантиомеров происходил перенос хиральности с имевшегося стереоцентра на возникающий – но строго зеркально. Если это не чудеса, то я не знаю, что такое чудеса. А теперь посмотрим на энантиоселективность, выражаемую разностью выходов продуктов с определенной конфигурацией нового центра, R минус S, или наоборот, в зависимости от того, чего больше. Сколько у нас (R)? Из одного трео и одного эритро 90+10 = 100% от половины, то есть 50%. А (S)? Из другого трео и другого эритро, смотрим по схеме, итого 90+10 = 100% от половины, то есть те же 50%. Разница равна нулю – нет энантиоселективности! Для каждого из энантиомеров субстрата есть, а в сумме, в конкретном экспериментальном результате реакции – нет! Это чертовски важно понимать, и не устану повторять – не забывайте, что оптическая активность и всё, что с этим связано проявляется только на уровне вещества, а не на уровне отдельных молекул. В этом сложность, сводящая многих с ума: на бумаге мы пишем уравнения реакций как будто на молекулярном уровне, и для многих вещей переход от молекул к веществу происходит незаметно – помножили в уме на число Авогадро и готово – какая разница, множитель постоянный, плевать, что такой громадный. Но в реальности это не простое умножение, а усреднение по огромному количеству молекул, каждая из которых находится в своем состоянии. И когда мы говорим про оптическую активность – мы говорим именно про вещество, огромное количество молекул, усреднение по этому множеству. Отдельные молекулы хиральны и в них происходит перенос хиральности в реакции. Но во множестве, в котором в начале не было оптической активности, она не могда появиться из ничего, и ровно так и получилось. С точки зрения вещества переноса хиральности не произошло! Рацемату – рацематово! Ни одна плоскость поляризации света не колыхнулась ни на сотую градуса, ни до, ни в ходе реакции, ни после!
Источник хиральности находится не только в субстрате, но и в реагенте
Возьмём тот же самый модельный субстрат с одним стереоцентром и прохиральным реакционным центром и сделаем ту же реакцию, но уже с реагентом (или катализатором, что одно и то же в этом контексте, да и вообще – реагент-некатализатор это в некотором смысле катализатор с TON = 1), имеющем элементы хиральности и введённом в реакцию в оптически активном виде. Последнее очень важно, так как с рацемической формой катализатора мы вряд ли получим что-то полезное. При этом рацемическая форма катализатора не может быть источником переноса хиральности только в том случае, если в субстрате нет своей оптической активности, а мы сейчас рассматриваем случаи, когда таковая есть. И в этом случае, если мы возьмём рацемат катализатора, то можем получить ситуацию, когда произойдёт расщепление энантиомеров катализатора на субстрате, и каждая форма реакционного комплекса будет реагировать с разными скоростями, потому что такие формы диастереомерны – но это чертовски сложная и совершенно непредсказуемая ситуация, с которой предпочитают не связываться, хотя может однажды повезти и получиться очень красивая работа. Но, скажем так, исследователи предпочитают не искать слишком много приключений на собственные шеи или иные части тел.
По той же причине почти никогда не будут применять оптически активный реагент (катализатор) к рацемическому субстрату. Энантиомеры имеют разные константы скоростей с такими реагентами, и с хорошей вероятностью смесь продуктов усложнится, вы потеряете даже диастереоселективность. Бывают, правда, случаи, когда константы скоростей настолько различны, что до некоторой, не очень большой глубина превращения можно считать, что реагирует только один. Такие ситуации называются кинетическим расщеплением энантиомеров, примеров много, но не так много, как обычных энантиомселективных реакций, Здесь обойдёмся без этого случая. Встретится, разберём.
Итак, чистый энантиомер субстрата и оптически активный рагент (катализатор). Первый вопрос: а был ли перенос хиральности без хирального реагента (катализатора)? Как мы убедились в первой части, вполне мог быть и часто бывает. Более того, эффективность этого переноса может быть разной с разными реагентами (катализаторами). Это вполне очевидно, потому что сама природа такого переноса определяется относительными энергиями переходных состояний на пути к двум диастереомерным продуктам, а они определяются конформациями и суммой взаимодействий в комплексе субстрат-катализатор. Взаимодействия эти в основном стерические, и тогда разные реагенты (катализаторы) дадут разные переходные состояния, с отличающейся разницей, и мы можем достичь и большей диастереоселективности и большей энантиоселективности, не применяя хирального реагента (катализатора). Но это такой же поиск, такая же кропотливая оптимизация, а нехиральные (или хиральные в оптически неактивной, рацемической форме) реагенты и катализаторы отлично могут быть намного дороже многих хиральных в оптически активной форме.
Но из этого буквально следует вот что: если субстрат оптически чист (в разумных пределах, никогда никто не требует 100%-ной чистоты, часто это и не нужно), то априорной разницы между хиральным и нехиральным реагентом (катализатором) нет потому что мы редко можем разделить, какая доля переноса хиральности обусловлена внутренним переносом от имеющегося стереоцентра, а какая идет от реагента. Вообще даже постановка такой задачи малость бессмыслена, потому что перенос хиральности отражает разницу энергий переходных состояний, а это уже комплекс из субстрата и реагента, и энергия такого комплекса естественно уже не может быть сведена к тому, что было до этого. Мы не можем разделить эти два типа переноса ни теоретически, ни экспериментально.
Но любая работа по энантиоселектиному синтезу всегда стремится достичь максимального энантиомерного избытка. Сейчас вы и не сможете опубликовать работу, если не получите результат, значимо превосходящий то, что уже достигнуто для субстратов и реакций того же типа. Реакций, предполагающих возможность достижения энантиоселективности много, и для каких-то уже давно решены все проблемы и делать больше нечего, хотя всегда остаются задачи упрощения, удешевления, достижения большей эффективности и т.д. А для каких-то дело идёт непросто и каждые следующие 10% прибавки энантиомерного избытка даются годами исследований и сотнями испробованных лигандов и катализаторов. Любой исследователь будет стремиться к максимуму, а для этого приходится просто пробовать разные катализаторы и лиганды, и в решении задач увеличения энантиоселективности в реакции субстрата, уже имеющего стереоэлементы и оптическую активность, когда сразу не получается добиться переноса хиральности от этих внутренних стереоцентров, почти всегда решение ищут в хиральных реагентах (катализаторах).
Более того. Представьте себе, что вы достигли очень хороших результатов с каким-то лигандом, у вас получился новый стереоцентр с очень хорошим избытком и вы счастливы, распеваете бодрую песню и готовитесь строчить статью в гламурный журнал. Но тут возникает вопрос, а можно ли получить точно то же самое, но с новым центром обратной конфигурации (например, вы получили очень хороший R, и теперь мечтаете о таком же хорошем S – при том, что все старые стереоэлементы остаются одинаковыми, то есть вы захотели уже тот диастереомер, который до этого получался как минорный). Получится ли это, если вы возьмёте тот лиганд, который дал вам отличный результат с R, но просто в виде энантиомера? Нет, не получится (чудеса бывают, но очень редко). Ужас в том, что вам придется искать новый лиганд, возможно совсем не похожий на тот, который уже отлично сработал, но на противоположной конфигурации. И не факт, что поиск приведёт к успеху.
Теперь разберём сам пример DO. Исходное представляет собой аллильный карбонат, типичный субстрат для аллильного замещения Цудзи-Троста. Кроме того, это оптически чистое соединение с двумя стерегенными центрами в непосредственной близости от реакционного центра, правда группы маленькие, но самая близкая – гидроксил. В результате реакции происходит смещение двойной связи и образуется новый стереогенный центр, а следовательно перед нами кандидат на диастерео- и энантиоселективность.
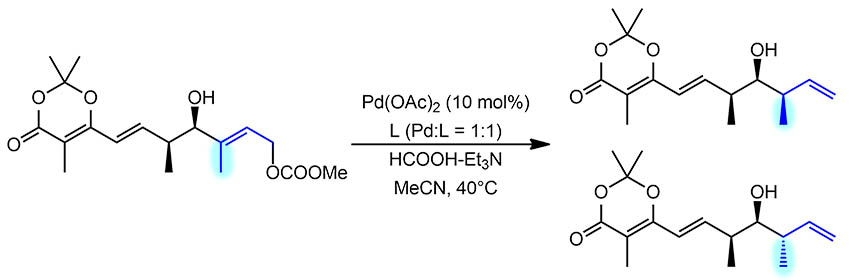
Вся дальняя часть молекулы нас не волнует. Механизм мы уже разобрали, просто повторим, что нульвалентный палладий (образуется прямо в реакционной смеси в пред-активации, скорее всего восстановлением Pd(2+) триэтиламином, но может и формиатом.Палладия немало, 10 моль%, на большое количество каталитических циклов авторы не рассчитывают, но это типично для стереоселективного синтеза, где главное – получить продукт с хорошей диастереомерной и оптической чистотой, а не экономия катализатора. Уменьшение загрузки, увеличение каталитических циклов обычно приводит к тому, что катализатор теряет селективность из-за деградации лиганда, предположительно источника хиральности. Итак, нульвалентный палладий расщепляет аллильный карбонат, цепляет гидрид из формиата, и переносит его на аллильную систему со смещением двойной связи. Почему здесь гидрид идет на более замещённый атом с аллильным сдвигом и образованием менее устойчивого терминального олефина, скзать непросто, но скорее всего это выгоднее именно по стерическим причинам, ведь палладий не сразу сходит вон, а перемещается дигапто-способом на двойную связь, и ему выгоднее оказаться дальше от стерически более затруднённой части молекулы.
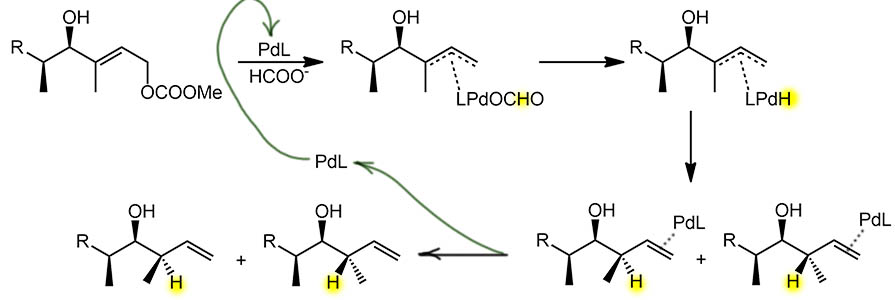 Посмотрим теперь на стереоселективность. Авторы заботливо снабдили нас данными по реакции без хирального лиганда в присутствии tBu3P, сильнодонорного объёмистого фосфина. Многие остальные лиганды тоже имеют диалкилфосфиновые центры, то есть авторы считают, в реакции нужен фосфин с высокой донорностью, скорее всего, это увеличивает реакционную способность гидридного лиганда, ведь реакция имеет нуклеофильную природу и никуда от этого не денешься. Так вот, с нехиральным фосфином диастереоселективность отсутствует полностью – соотношение
Посмотрим теперь на стереоселективность. Авторы заботливо снабдили нас данными по реакции без хирального лиганда в присутствии tBu3P, сильнодонорного объёмистого фосфина. Многие остальные лиганды тоже имеют диалкилфосфиновые центры, то есть авторы считают, в реакции нужен фосфин с высокой донорностью, скорее всего, это увеличивает реакционную способность гидридного лиганда, ведь реакция имеет нуклеофильную природу и никуда от этого не денешься. Так вот, с нехиральным фосфином диастереоселективность отсутствует полностью – соотношение 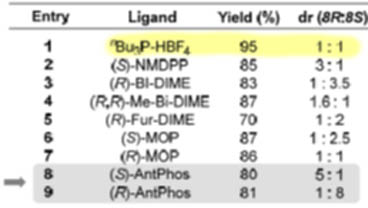 диастереомеров 1:1, то есть имеющиеся стереоцентры вообще никак не участвуют, ни стерикой, ничем ещё. Хорошо, бывает, гидрокcил маленький и никак с палладием, на котором висит большой фосфин, не взаимодействует. Ну, тем яснее роль хирального лиганда. Авторы перебирают целую пачку лигандов, все монодентатные донорные фосфины, многие относятся к типу, предполагающему атропо-изомерию, но здесь она, видимо совсем не работает. Фосфины дают диастереоселективность, но большинство небольшую, некоторые вообще никакую. Понять почему крайне трудно, и подавляющее большинство исследователей стереоселективного синтеза такой задачи не ставят – это потребовало бы моделирования механизма и переходных состояний, работа тяжелая и крайне неблагодарная, потому что мы не совсем точно представляетм себе происходящее – все наши механизмы довольно приблизительны и имеют много допусков. Ограничимся наблюдением, что можно получить преобладание обоих диастереомеров, и это тоже скорее всего является следствием неучастия стереоцентров молекулы в стереодискриминации путей реакции – иными словами, реакция идет практически так же, как если бы их и не было – в режиме энантио-дискриминации с переносом хиральности исключительно от лиганда. Но – не всё так просто, химия не дает возможности строить кристально ясные схемы. Если бы это было совсем чисто так, то энантиомерные катализаторы давали бы одинаковую энантиоселективность, – энантиомеры продукта получались бы с приблизительно одинаковыми соотношениями (естественно, в пределах точности эксперимента, видимо, очень небольшой – большинство практикующих органиков вообще понятия не имеет про необходимость добиваться статистически достоверных результатов и каждый опыт ставят один раз, статистическая достоверность таких результатов слабо отличается от нуля). В таблице есть два примера применения энантиомеров одного лиганда – очень хорошо известный лиганд MOP, единственный в списке использованных, имеющий стереогенную ось (по другой интерпретации – стереогенную плоскость) и обязанный хиральностью атропоизомерии. Видим, что (R)-форма лиганда просто провалилась по стереоселективности, хотя реакция идёт не хуже чем с остальными лигандами – в общем, мы видим, что она со всеми лигандами идет приблизительно одинаково, потому что серьёзно сравнивать выход в 70% и 96% могут только люди, прогулявшие на младших курсах семинары и лекции по матстатистике. А (S)-форма таки дает вполне заметную селективность. Такая асимметрия может иметь только два объяснения – либо результаты плохие и кто-то что-то напутал, либо всё таки имеющийся стереоцентр участвует в стереодикриминации, но это участие очень чувствительно к структуре лиганда. Ещё одно любопытное наблюдение над результатами авторов – относительно неплохое выступление одного из самых старых и примитивных хиральных лигандов, неоментилдифенилфосфина (NMDPP), полученного из природного ментола, одного из самых дешевых и легкодоступных оптически чистых соединений, и давно заклеймлённого в асимметрическом синтезе как малоэффективный, потому что просто стереоцентры в лиганде очень слабо рассимметризуют пространство вокруг реакционных центров. Вот так она устроена, химия – идут годы и десятилетия, появляются тысячи новых изощренных катализаторов и лигандов, десятки новых концепций стереодифференциации, но появляется очередная работа, где плевать все хотели на все эти красивые принципы, просто берут десяток доступных лигандов без всякого обоснования, и получают очередную таблицу оптимизации, где всё вместе, и не поймёшь, зачем были все эти исследования и концепции. Но другой химии у меня для вас нет – будете работать сами, попробуйте планировать свои исследования умнее и в соответствии с новейшими воззрениями.
диастереомеров 1:1, то есть имеющиеся стереоцентры вообще никак не участвуют, ни стерикой, ничем ещё. Хорошо, бывает, гидрокcил маленький и никак с палладием, на котором висит большой фосфин, не взаимодействует. Ну, тем яснее роль хирального лиганда. Авторы перебирают целую пачку лигандов, все монодентатные донорные фосфины, многие относятся к типу, предполагающему атропо-изомерию, но здесь она, видимо совсем не работает. Фосфины дают диастереоселективность, но большинство небольшую, некоторые вообще никакую. Понять почему крайне трудно, и подавляющее большинство исследователей стереоселективного синтеза такой задачи не ставят – это потребовало бы моделирования механизма и переходных состояний, работа тяжелая и крайне неблагодарная, потому что мы не совсем точно представляетм себе происходящее – все наши механизмы довольно приблизительны и имеют много допусков. Ограничимся наблюдением, что можно получить преобладание обоих диастереомеров, и это тоже скорее всего является следствием неучастия стереоцентров молекулы в стереодискриминации путей реакции – иными словами, реакция идет практически так же, как если бы их и не было – в режиме энантио-дискриминации с переносом хиральности исключительно от лиганда. Но – не всё так просто, химия не дает возможности строить кристально ясные схемы. Если бы это было совсем чисто так, то энантиомерные катализаторы давали бы одинаковую энантиоселективность, – энантиомеры продукта получались бы с приблизительно одинаковыми соотношениями (естественно, в пределах точности эксперимента, видимо, очень небольшой – большинство практикующих органиков вообще понятия не имеет про необходимость добиваться статистически достоверных результатов и каждый опыт ставят один раз, статистическая достоверность таких результатов слабо отличается от нуля). В таблице есть два примера применения энантиомеров одного лиганда – очень хорошо известный лиганд MOP, единственный в списке использованных, имеющий стереогенную ось (по другой интерпретации – стереогенную плоскость) и обязанный хиральностью атропоизомерии. Видим, что (R)-форма лиганда просто провалилась по стереоселективности, хотя реакция идёт не хуже чем с остальными лигандами – в общем, мы видим, что она со всеми лигандами идет приблизительно одинаково, потому что серьёзно сравнивать выход в 70% и 96% могут только люди, прогулявшие на младших курсах семинары и лекции по матстатистике. А (S)-форма таки дает вполне заметную селективность. Такая асимметрия может иметь только два объяснения – либо результаты плохие и кто-то что-то напутал, либо всё таки имеющийся стереоцентр участвует в стереодикриминации, но это участие очень чувствительно к структуре лиганда. Ещё одно любопытное наблюдение над результатами авторов – относительно неплохое выступление одного из самых старых и примитивных хиральных лигандов, неоментилдифенилфосфина (NMDPP), полученного из природного ментола, одного из самых дешевых и легкодоступных оптически чистых соединений, и давно заклеймлённого в асимметрическом синтезе как малоэффективный, потому что просто стереоцентры в лиганде очень слабо рассимметризуют пространство вокруг реакционных центров. Вот так она устроена, химия – идут годы и десятилетия, появляются тысячи новых изощренных катализаторов и лигандов, десятки новых концепций стереодифференциации, но появляется очередная работа, где плевать все хотели на все эти красивые принципы, просто берут десяток доступных лигандов без всякого обоснования, и получают очередную таблицу оптимизации, где всё вместе, и не поймёшь, зачем были все эти исследования и концепции. Но другой химии у меня для вас нет – будете работать сами, попробуйте планировать свои исследования умнее и в соответствии с новейшими воззрениями.
Самым лучшим лигандом оказался монофосфин,сокращённый как AntPhos, с кокетливым намёком на муравьёв, но понятно, что от названия антрил, тоже вроде бы построенный на атропоизомерной основе. Но это обманка, это в действительности самый простой хиральный фосфин со стереогенным атомом фосфора, а для пущей конфигурационной устойчивости фосфор встроили в гетероцикл. У этого лиганда нет стереогенной оси, хотя может показаться, что она должна быть – но мы не можем представить две формы, отличающиеся углом скручивания – влево или вправо – относительно этой оси, потому что один из таких воображаемых диастереомеров будут иметь трет-бутильную группу направленную внутрь угла раствора между верхним и нижним кольцом, что невозможно по стерическим причинам. При этом пери-протоны антрильной системы не дают верхнему кольцу провернуться на обратную сторону. Итак, это просто лиганд с одним стереогенным центром, но с очень объёмистым асимметрическим окружением этого центра. Конфигурация атома фосфора определяется по обычной системе, при этом неподеленная пара считается самым младшим заместителем. Для (S)-формы все совсем просто – нарисовали стрелочку по убыванию страшинства и готово. А вторая форма по определению (R), и определять ничего не надо. Вот у этого лиганда получилось, причем были получены оба энантиомера и в реакции они дают противоположную конфигурацию стереогенного центра, но с значительным различием – то есть и здесь мы видим интерференцию с имеющейся в субстрате хиральностью, причем очевидно, что одному направлению реакции она помогает, а другому мешает.
В случае (S)-AntPhos основной продукт имеет (R)-конфигурацию нового центра, диастереоселективность измеряется отношением 5:1, что приблизительно соответствует соотношению диастереомеров 83:17, и тогда мы можем оценить меру энантиоселективности, энантиомерный избыток ee = 83-17 = 67% (c учетом округления). Не густо. В случае (R)-AntPhos основной продукт имеет (S)-конфигурацию нового центра, диастереоселективность заметно выше, 8:1, или около 89:11, и энантиомерный избыток ee = 89-11 = 77% (с учётом округления). Напомню еще раз, что в случае образования диастеромеров энантиомерный избыток не означает преобладания одного энантиомера над другим, другого энантиомерна просто вообще нет (продукты – это диастереомеры SSR и SSS, а энантиомером были бы или RRR или RRS, ну и откуда им взяться?), так как все старые стереоцентры не изменили конфигурацию. Но это мера именно энантиоселективности, затрагивающая только тот центр, который возник заново в результате реакции которая привела к преобладанию одной конфигурации над другой.
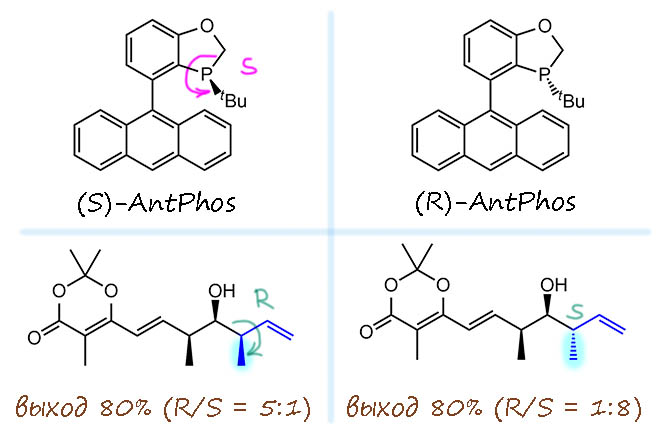
Итак, мы видим существенную разницу между результатами с двумя энантиомерами лиганда. Такой разницы не было бы, если бы в субстрате не было бы других стереоцентров, или если бы имеющиеся стереоцентры вобще не влияли бы, как можно было бы решить из отсутствия диастереоселективности и энантиоселективности в эксперименте с оптически неактивным лигандом.
DH
Очередной подход к веществам из тисса головчатого, цефанолидам. На этот раз еще одна китайская бригада под руководством профессора Ху Сяндуна из Северо-западного университета города Сиань. Когда вам в следующий раз будут пудрить мозги насчет того, что Китай – противник, конкурент, или даже соперник США, обратите внимание на то, как тщательно, почти раболепно копируют китайцы всякие знаковые американские явления. Вот и университет назвали Северо-западным, почти точно скопировав модную в США манеру сокращать названия своих знаменитых университетов. А в Штатах Северо-западный университет близ Чикаго – один из мощнейших и знаменитейших, и никто не переспрашивает, когда слышит про какой-то северо-западный университет или даже просто северозападный, the Northwestern, на северо-западе чего тот находится и где это вообще – все и так знают, там по берегу озера Мичиган нобелевские лауреаты толпами ходят взад-вперёд и не могут разойтись, так их много. Вот и китайцы тоже, создавая несколько десятилетий назад с нуля науку и образование, создали сеть больших университетов на манер американских, и не удержались от того, чтобы не назвать один Северо-западным, хотя город Сиань находится скорее в центре Китая, впрочем действительно западнее большинства других университетских центров. Нобелевских лауреатов, правда, пока не завезли, но какие их годы. Соперник и конкурент это не тот, кто копирует. Тот кто копирует называется словами ученик, подражатель. Да, при этом часто произносятся всякие задиристые речи, а иногда даже и раздается аккуратное бряцание чем-то смахивающим на оружие, впрочем тщательно поставленным на предохранитель или же вовсе незаряженным – но это только для коррекции самооценки, не более. А на деле китайцы скромно и усердно учатся, не пропуская ни единого слова учителей. Науку в Китае построили строго по образцу американской, да и работают в ней те, кто хорошенько выучился у лучших профессоров за океаном. Отсюда и огромные успехи в очень сложных областях науки, достигнутые за кратчайшее историческое время – всего пара десятилетий. Посмотрим на еще один синтез, сделанный современными китайскими учеными.
В данном задании разбирается пример применения реакции Посона-Кханда. Это ключевая стадия синтеза. Мы уже по другим примерам синтезов цефанолидов знаем, что в этих соединениях есть система циклов, которые и нужно строить. В данном случае реакцией Посона-Кханда решили построить два цикла. Авторы утверждают, тчо использовали диастереоселективную реакцию Посона-Кханда, и в этом месте у нас сначала может возникнуть недоумение – а что там, в этой реакции диастереоселектвного – ведь это циклизация из олефина, ацетилена и карбонила. Диастереоселективность предполагает возникновение одного нового стереоэлемента в молекуле, уже имеющей стереоэлементы. Или двух новых стереоэлементов, тогда в молекуле могут быть, а может и не быть “старых” стереоэлементов. Хорошо, тогда мы видим, что стереохимия в этой реакции может возникнуть только из олефина, если олефин хотя бы дизамещенный, то в циклизации возникают два новых стереоцентра. Но поскольку циклизация с образованием не очень больших циклов по определению син-процесс, значит мы назовем ее син-диастереоспецифичной, и при отсутствии в одной из циклизующихся молекул других стреоэлементов, мы могли бы получить только энантиоселективность, если можно было бы придумать, как перенести хиральность с реагента или катализатора – здесь мы сразу видим проблему, потому что в классической реакции Посона-Кханда катализатор, карбонил кобальта, простая нехиральная молекула и куда там можно было бы воткнуть оптически активный анциллярный лиганд априори непонятно. Оставим это пока – в данном задании этой проблемы нет. И мы будм получать рацемический продукт циклиизации, но с сохранением взаимного расположения заместителлей из олефина.
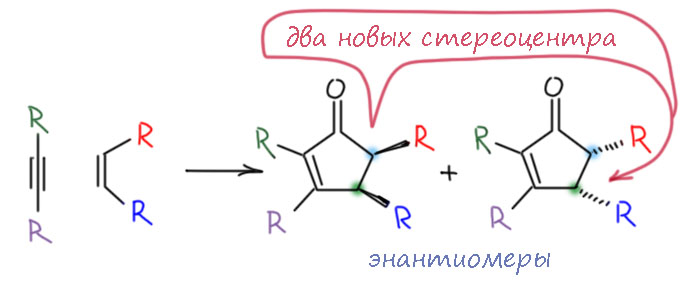
Но теперь представим себе, что в олефине уже есть стереоэлементы. Тогда относительно этих элементов мы можем получить (или не получить) диастереоселективность. Когда стереоэлементов (почти всегда это стереоцентры, но мы уже научились так же лихо орудовать со стереоплоскостями и стереоосями, и поняли, что принципиальной разницы нет) больше двух, в реакции может быть несколько типов диастереодискриминации – реакция может быть, например, диастереоспецифична по конфигурации фрагмента, образованного из двойной связи; и диастереоселективна относительно взаимного расположения этого фрагмента и других стереоэлементов.
Обратимся теперь к структуре задания, чтобы увидеть это на конкретном примере. Начнем с того, что представим себе, как образуются циклы на уже готовом бициклическом каркасе, оборвав все лишнее и оставив только самое основное. Мы видим, что цепь с ацетиленом, прикреплена к углероду в голове моста, и поэтому система совершенно симметрична – плоскость симетрии проходит через мост с двойной связью. Для наглядности нарисуем еще и ньюмена, выбрав первую связь цепи. Тогда по этой связи у молекулы быдут два совершенно одинаковых по энергии, но энантиомерных конформера. Циклизация поэтому из каждого конформера будет иметь одинаковую энергию активации и одинаковую константу скорости – в результате мы получим рацемическую смесь. Эти два продукта энантиомерны, хотя это не сразу можно понять, просто посмотрев на структуры, ведь они нарисованы под углом и кажутся разными. Но нет – они отражаются друг в друга в зеркале. Это случай, уже вполне рассмотренный на первой схеме – циклизация может происходит с той и другой стороны плоскости двойной связи.
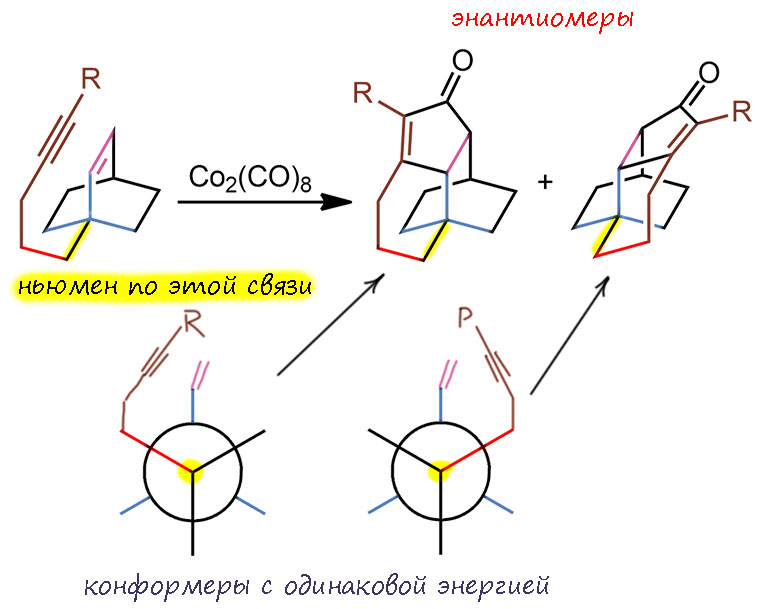
А теперь добавим то, что есть в реальной структуре – бицикл там не просто углеводородный, а гетероциклический. Я перерисовал проекции так, чтобы самые важные события происходили на переднем плане, где это виднее. Даже неважно какой конкретно, могли просто быть заместители, но симметрия тут же разрушается. Плоскости симметрии больше нет, конформеры больше не одинаковы по энергии, стерическое отталкивание с одной стороны сделало их разными. Более выгодный то, который со стороны, где нет заместителя. Как всегда в таких “проволочных” формулах кажется, чьл там не может быть серьёзных препятствий и заместитель находится далеко от вращающегося хвоста, на котором болтается ацетилен. Но это иллюзия, на самом деле объём атомов значителен и отталкивание досточно для дискриминации конформеров. Продукты тоже больше не энантиомеры, это диастереомеры, потому что после уничтожения плоскости симметрии в исходном бицикле атомы в голове моста стали стереоцентрами.
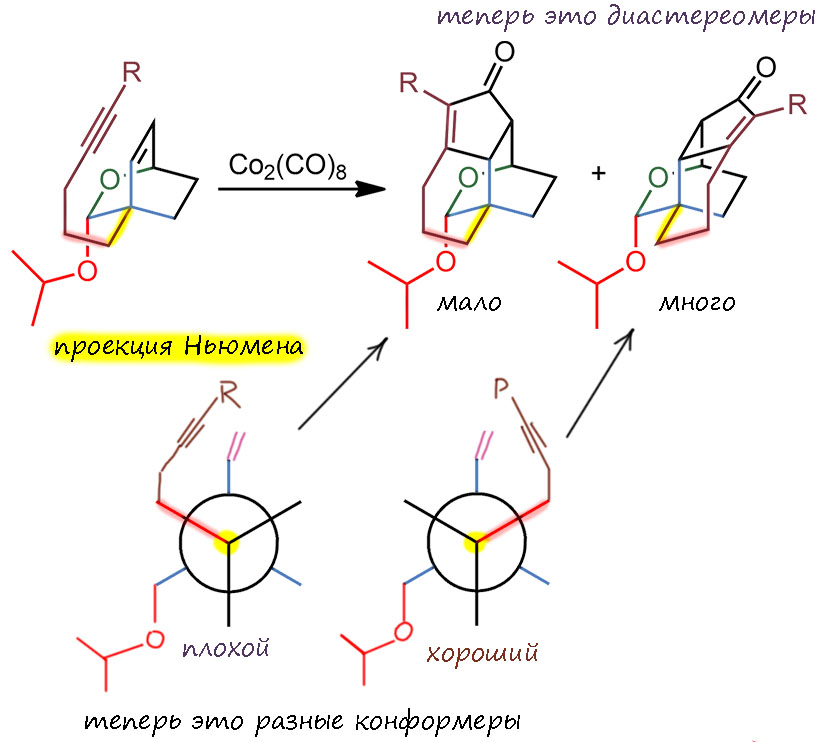
Теперь можно окончательно нвесить всё, что есть в работе, и нарисовать схему реакции. Мы видим, что диастереоселективность выражается неплохой ведличиной 10:1 в пользу нужного диастереомера – хорошая, хотя и не самая высокая селективность. Но тут как раз и всплывает то, что заместитель все же довольно далеко, и дискриминация между конформерами, а точнее переходными состояниями (вспомните, что такое кинетика Кёртина-Гаммета) не очень большая, но, в принципе, для задач синтеза достаточная. Дальше можно дочистить хроматографией. Реакция некаталитическая, и хотя Посона-Кханда можно делать каталитически, но в сложных синтезах это редко делают – карбонил кобальта штука дешёвая.
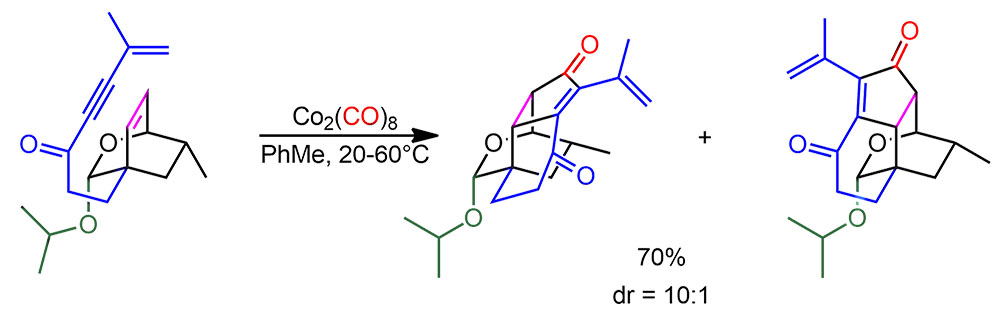
И последний вопрос, который прямо напрашивется. А это всё оптически активно? И да и нет. Насколько можнл понять из статьи, вся эта работа сделана на рацемическом исходном. И это никак не влияет на результаты, потому что как мы уже выяснили диастерео- и энантиоселективность не зависят друг от друга: диастереоселективность можно отлично продемонстрировать на рацемате, а потом сделать начисто на оптически чистом исходном, при этом всё, что было установлено про диастереоселективность, никак не изменится. Китайские исследователи вроде бы именно так и поступили. Экономные ребята.