
Карбены Фишера
Первый карбеновый комплекс переходного металла получил хорошо нам знакомый Отто Фишер в 1964 году (Fischer E. O., Maasböl, A. Angew. Chem., Int. Ed. 1964, 3, 580). Фишер явно озадачился вопросом – а насколько может быть похожа карбонильная группа в органике на карбонильный лиганд в комплексах. И провел самую обычную для карбонильной группы реакцию – с нуклеофилом, с литийорганикой, взяв для начала карбонил вольфрама. Выбор металла, как оказалось, был совершенно гениален. У этого великого ученого было какое-то потрясающее чутьё на открытия, причём надо хорошо понимать, что дело происходит в 1960-х и это самое-самое начало осмысленной химии переходных металлов, но очень многое ещё совершенно непонятно и не сложилось. Это мы сейчас всё уже знаем, лихо анализируем и подтверждаем – да, правильно, удачный выбор. Но тогда-то было не так. И объянить очередное попадание Фишера в точку можно только одним – некоторые химики руководствуются не только знаниями (знаний там через край, но уровень знаний соответствует началу развития области, и с этим ничего не поделаешь – в статьях Фишера вы не найдете теоретических обобщений, они в осоновном все довольно типично немецкие, даром что он всю свою карьеру продолжал писать по-немецки в немецкие журналы), но даже скорее интуицией, они чувствуют вещества, знают все повадки; эти люди всю свою жизнь провели в лабораториях и лично участвовали в важных экспериментах, ежедневно погружались в работу сотрудников, и так развивали это сверхестественное чутьё вещества. Безусловно, работу ученых такого уровня как Отто Фишер вполне уместно назвать искусством.
Именно металлы 6-й группы идеальны для карбенов этого типа, и мы сейчас это увидим, потому что это металлы ровно из серединки, а здесь невероятно важен баланс back-donation и обычной координационной связи, и это ровно то, что можно ожидать от 6-й группы, в общем-то не ранней, и не поздней.
Вернемся к реакции. Реакция прошла, образовался анионный аддукт, вполне стабильный, его удалось осадить в виде кристаллического осадка. В растворе его можно протонировать, а затем действием диазометана превратить в метилированное производное. Фишер назвал это первым карбеновым комплексом металла. Мощь мысли Фишера впечатляет. Если вы думаете, что карбены это в общем-то штука понятная, то может быть сейчас это более-менее так, но история осмысленной химии карбенов начинается в 1950-х, и сначала надо было просто разобраться с простыми карбенами. И химия переходных металлов возникла тогда же, и это мы уже знаем, особенно если послушали вводную лекцию – и Фишер был одним из отцов основателей, и за 10 первых лет он уже столько всего сделал, что голова кружится. Но он ненасытен, и теперь решил соединить одну новую химию с другой, столь же новой. Получилось! Химия карбенов всех очень увлекла в те годы и в неё бросились многие, так что через 10 лет после начала уже писали книжки. Фишер явно не искал карбены, он именно просто присоединил нуклеофил к карбонильному лиганду, и явно был удивлен, что получилось нечто стабильное, но не упустил – да это же первый комплекс карбена!
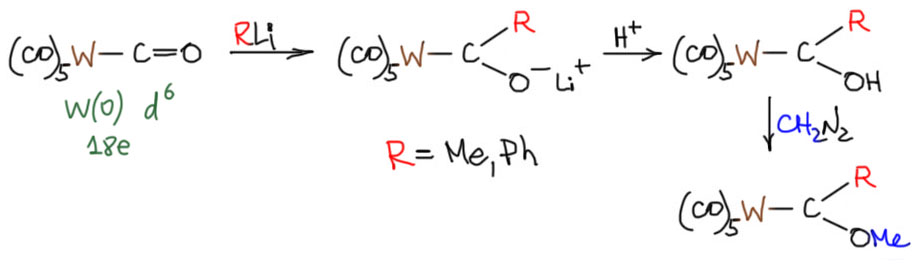
Почему Фишер решил, что это карбен. Да очень просто – на углероде всего две группы, то есть, если отсоединить этот лиганд от металла, мы увидим карбен. Что это будет за карбен? На этой схеме их три, и первый аддукт, и протонированная форма, и конечный комплекс, уже совершенно устойчивый и легкий в обращении. Если мы бы имели такой карбен отдельно, то что бы это было? Это вполне хорошо известно. Карбены с одной мезомерно-донорной группой это короткоживущие очень активные частицы, но относительно самого простого кабена они во-первых, стабилизированы и во-вторых в основном состоянии синглетны и не имеют проблем с триплетным состоянием. Но, ещё раз повторю, это короткоживущие активные частицы, которые можно наблюдать спектроскопически или при низких температурах в матрицах или в вакууме. А в комплексе этот карбен совершенно стабилен – в этом месте мы не впадаем в маразм, и понимаем, что в комплексе это уже не карбен в полном смысле слова, а такой лиганд.
Карбеновые комплексы такого типа стали называть карбеновыми комплексами или просто карбенами Фишера. Будьте осторожны с этим популярным термином – комплексы, называемые карбенами Фишера не являются карбенами, это координационные соединения разных переходных металлов с лигандами, которые были бы карбенами, если бы их можно было оторвать от металла, но оторвать нельзя – большинство таких карбенов в свободном состоянии совершенно неустойчивы и быстро превращаются в устойчивые соединения за счет секстетных перегруппировок. Но мы тоже будем для краткости называть такие комплексы карбенами Фишера.
Новые комплексы очень всем понравились и их стали интенсивно исследовать. Сам Фишер со своей лабораторией долго был впереди, но понемногу набилось много другого народа. Карбеновые комплексы Фишера были обнаружены и для других металлов, в основном из второй половины рядов. Для самых ранних переходных металлов они не свойственны, потому что там очень силен эффект back-docation, который погасит электрофильность полностью и даже из такого карбена сделает Шрока. Слишком в сторону подних тоже плохо – там у металла обычно слишком много электронов, и они тоже будут здорово гасить электрофильность. И за карбенами Фишера оказалась закреплена 6-я группа: хром, молибден, вольфрам. Причем на металле должно быть несколько карбонилов, не обязательно все, но и не одинЗдесь надо понять, что под карбенами Фишера понимают далеко не любые карбеновые лиганды. И здесь есть некоторая историческая проблема. Фишер открыл первый карбеновый комплекс – в этом состояла цель его работы, даже если в первый момент результат и оказался неожиданным. И таким образом показал, что из карбенов получаются отличные лиганды. Дальше и сам Фишер и другие исследователи стали открывать карбеновые комплексы в больших количествах, от самых простых до весьма сложных. И увидели, что при вариации и металла, и других лигандов на металле, и заместителей на карбене и даже шире – структуры карбена – и электронная структура, и свойства, и реакционная способность карбеновых комплексов в целом и карбеновых лигандов в частности изменяются невероятно широко – они все разные, у них разные функции, разные реакции. И тогда за термином “карбены Фишера” оставили очень узкую область – и очень важно понять, что важно все. Тот же карбен на другом металле может не быть карбеном Фишера. Другой карбен на тех же карбонилах хрома, молибдена, вольфрама не будет карбеном Фишера.
Иногда, особенно в торопливо-поверхностных курсах можно увидеть такое понимание карбенов в химии переходных металлов – есть два типа кабренов или карбеновых комплексов: карбены Фишера и карбены Шрока, и у них такие-то и такие-то особенности. Возникает ощущение, что любой карбен должен быть или одним или другим, и например, в метатезисе, где много карбенов, есть одни и другие. Но это заблуждение. В метатезисе, например, вообще нет карбенов Фишера. Карбены Фишера – это очень узкая, но и очень интересная область. У них огромное количество применений в органике, и при этом – почти исключительно стехиометрические реакции, почти совсем нет катализа.
Главная особенность карбенов Фишера – исключительно тонкий баланс взаимодействий. Back-donation нужен, без него комплекс не будет должным образом стабилизирован, и не будет легко получаться и быть удобным для исследований и применений. А одна из важнейших фишек карбенов Фишера – необычайная легкодоступность. Но back-donation не должен быть слишком силен, тогда мы потеряем многие особенности этого типа лигандов; поэтому у нас (то есть у Фишера, но примазаться всегда приятно) есть металл в низковаленлентной форме, готовый к back-donation. Но на металле висит еще пачка карбонилов, которые как голодные псы растаскивают эти электроны, и держат back-donation в очень умеренной дозе.
Общий признак карбенов Фишера – мезомерные доноры на углероде, обычно один. Чаще всего это кислородная группа, алкокси, ацилокси или что-то похожее. Азот тоже бывает, но намного реже, потому что слишком большой донорный эффект тоже нежелателен, потом поймём почему. Ещё бывает сера, но это уже почти экзотика. Карбеновый лиганд связан с металлом координационной связью, на что уходит неподеленная пара. Мы знаем, что у карбенов есть еще вакантная орбиталь, и в карбеновых комплексах эта орбиталь обычно становится участником back-donation. А здесь еще и мезомерный эффект от донорного заместителя. Сначала про back-donation: оно не слишком сильно влияет, потому что со стороны металла есть другие лиганды и это очень часто карбонилы, которые и забирают все ресурсы back-donation. Мезомерный донор с другой стороны безусловно как-то участвует, но полностью закрыть не может. Иными словами, мы получаем некий аналог карбонильного углерода, электрофильный центр. Аналогию с карбонильным соединением можно провести дальше – на α-углероде может быть атом водорода, тогда он становится весьма кислым, намного более кислым, чем в карбонильных соединениях.
Стоит плучше разобраться в том, как устроены такие комплексы, и как это можно изобразить на бумаге. Обычно прибегают к мезомерии и граничным структурам, только надо никогда не забывать, что у переходных металлов не всё так просто и однозначно, как у обычной органики. Вот,например, как это делает Карл Хайнц Дёц, ученик самого Фишера и учёный, посвятивший всю жизнь изучению карбенов Учителя и немало в этом преуспевший. Ему потребовалось три граничные структуры.
В первой и самой распространённой, обычно и использующейся как основная, связь металла с углеродом показана как двойная. В этом месте мы сильно напрягаемся, потому что вторая черта здесь изображает именно back-donation, а мы до сих пор избегали явного графического отображения этого эффекта в структурах, и подробно обсуждали почему – потому что это эффект переменной величины, обычно работающий не как полноценная связь, а как такой инструмент тонкой настройки. Очень важно понимать, что карбен это лиганд L-типа, и изображение двойной связью это никак не меняет. Если угодно, это можно считать старыми добрыми валентностями. Углерод четырехвалентен, ему положены четыре чёрточки-валентности, вот мы их и рисуем. И есть ещё один подход, прорисовавшийся в последние годы вместе с принятием ещё одного типа лигандов – Z-типа, специально для back-donation. Тогда карбен можно считать лигандом LZ-типа, что весьма удобно, так как Z не изменяет ни счёт электронов, ни степень окисления, ни счет валентных электронов. Вторая граничная структура как будто разгоняет эту вторую связь, смещая её к металлу, на металле появляется формальный отрицательный заряд . на углероде положительный, и видно, что углерод имеет три связевые черты и это карбокатионный центр. В этой структуре все правильно, хотя и необычно, но металл сохраняет степень окисления ноль, потому что – и это довольно тяжёлое испытание для нормального человека, но у меня для вас нет другой координационной химии – в граничной структуре тип лиганда может меняться и здесь он стал Х-лигандом. Почему я так решил? Да посмотрите на него сами в этой структуре и убедитесь, что это одновалентный остаток, а не нейтральная частица. Тогда степень окисления металла в этой структуре -1+1=0. Третья граничная структура очевидно получается из второй и затрагивает только органическую часть.
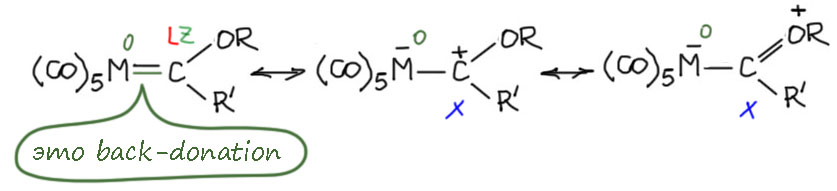
Теперь посмотрим на химию карбенов Фишера. Их главная, но не единственная реакционная способность вполне выражается приведенными граничными структурами – это электрофильные реагенты с самым главным реакционным центром на “карбеновом” углероде. Этот углерод является еще и акцептором в смысле электронных эффекотов в органической части молекулы. Во многих реакциях переходгный металл с остальными лигандами остается на месте и участует в реакциях только за счет стабилизации промежуточных продуктов, в которых сохраняется электронодефицитный атом углерода.
Реакции карбенов Фишера
Карбены Фишера смахивают на карбонильные соединения
Эта аналогия очень хорошо читается, но относиться к ней надо аккуратно, не забывая про металл с лигандами, который здесь работает как очень своеобразный заместитель, полного аналога которому мы в обычной органической химии не найдём. И сейчас прямо на разных реакциях мы увидим это разнообразие и аналогии.
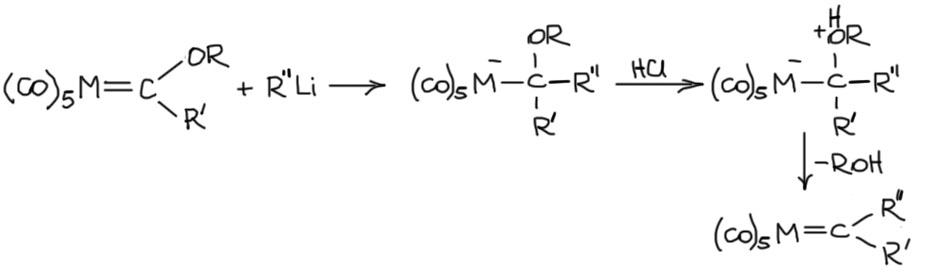
Первую реакцию с нуклеофилом сделал сам Фишер. Это была реакция с литийорганикой. Напомню только, что и сам карбен Фишера получен из реакции карбонила шестой группы с литиорганикой. Получается, что резервы электрофильности, заложенные в карбонильном комплексе хрома, молибдена или вольфрама так просто исчерпать не удалось. Итак, присоединяем эквивалент литийорганики, это очень удобно сделать, использовав одну из граничных структур, ту, которая подчёркивает электрофильный характер углерода. Собственно можно и другие взять, но раз уж мы так разобрались в том, как устороены эти структуры, грех не пользоваться результатами собственного трудолюбия. Видим, что образовался аддукт, прямо бешено похожий на обычный тетраэдрический аддукт у карбонильных соединений и вполне устойчивый. Металл при этом не изменяет степени окисления.
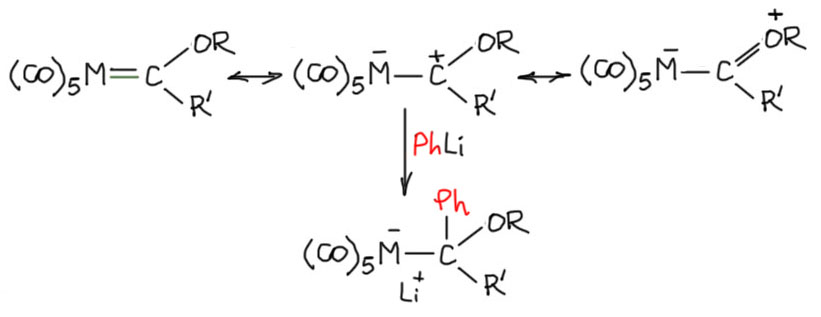
Такое впечатление, что металл сидит на двойной связи как карбонильный кислород, и присоединение нуклеофила точно следует паттерну присоединения нуклеофилов к карбонильной группе. Но это не совсем так, хотя аналогия красивая и во многом работающая. Фактически это похоже на лигандный обмен нейтрального L-лиганда на отрицательный X-лиганд. Формальный заряд принадлежит всему комплексу. По-другому можно представить как карбен присоединяет нуклеофил и превращается в карбанион. И новый комплекс это формально продукт лигандного обмена нейтрального L-лиганда на анионный X-лиганд.
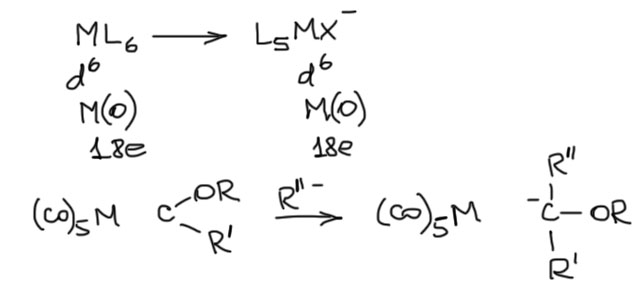
Аддукт устойчив, но алкокси-группу можно убрать, это впервые сделал сам Фишер обычной кислотой, после в ход пошли более мягкие и селективные кислотные реагенты, особенно удобным оказался ТМС-трифлат. То, что при этом образуется, очень легко опознать как новый карбеновый комплекс. Но хоть этот комплекс и получил впервые Фишер, это не карбен Фишера. Это просто карбеновый комплекс, который, возможно, тоже для чего-то нужен, но он не имеет свойств электрофильного карбена, напоминающего карбонильное соединение. Но способ генерации новых карбеновых комплексов действием литийорганики с последующим элиминированием алкокси-группы остался.
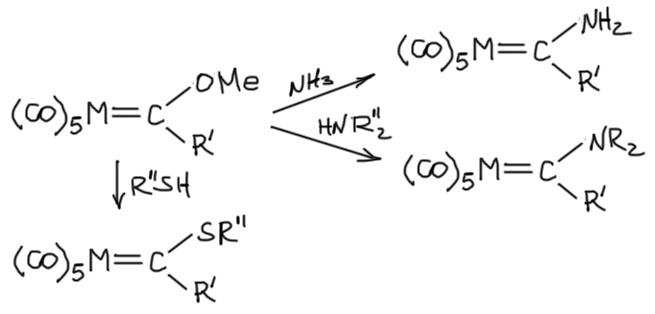
Еще проще идут реакции с простыми нуклеофилами: аммиаком, аминами, тиолами. Просто при комнатной температуре без добавления других реагентов сразу образуются новые карбены Фишера (Fischer, E. O.; Klabunde, U. J. Am. Chem. Soc. 1967, 89, 7141). А вот это всё типичные карбены Фишера, и свойства у них очень похожи. Есть даже такая аналогия – карбены Фишера рассматривают как такие проихводные карбоновых кислот: сложные эфиры, амиды, тиоэфиры.
CH-кислотность и аналогия с енолизуемыми карбонильными соединениями
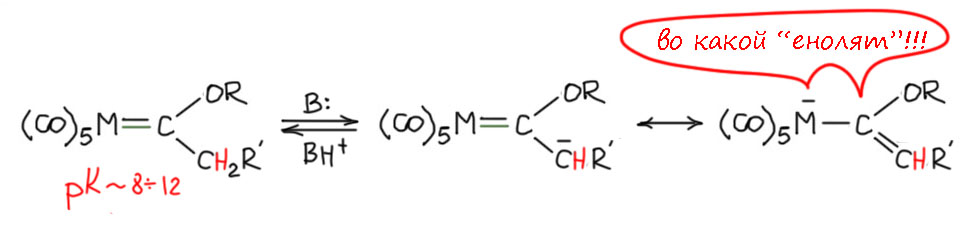
Раз есть аналогия с карбонильными соединениями в электрофильности, нет ли такой же аналогии в CH-кислотности α-протонов относительно карбенового углерода. Есть, и впервые её обнаружили на дейтерообмене с метилатом натрия в дейтерометаноле (Kreiter C. G. Angew. Chem., Int. Ed. 1968, 7, 390). Кислотность этих протонов оказалась весьма велика. В литературе чаще всего фигурирует величина рК 8 в водном растворе, но есть и более осторожные мнения, указывающие на методические проблемы в ранних измерениях и на то, что кислотность всё же немного пониже, но это всё равно что-то около 12 единиц. То есть это нечто близкое к малоновому эфиру, ну или если верить той цифре можно сказать так: по кислотности эти соединения находятся рядом с нитроалканами. Иными словами для оратимого депротонирования вполне достаточно аминов. Вспоминаем химию карбонильных соединений и то, что еноляты бывают равновесные и стехиометрические. Для первых как раз нужно основание сравнимой силы, а для вторых – намного более сильное основание. Сопряжённое основание можно нарисовать граничными структурами, и тогда мы увидим вроде бы совсем удивительное – как будто бы металл играет ру роль, котороую в обычных енолятах играет кислород карбонила. Но опять предостерегаю от слишком буквальной аналогии – здесь речь идёт скорее не о таком же смещении электронной плотности, а о вариации эффекта back-donation: после образования “енолята” карбеновый атом углерода получает плотность от анионного центра, и больше не нуждается в back-donation; и этим минусом на металле мы и обозначаем это – пара для back-donation не нужна и осталась на металле.
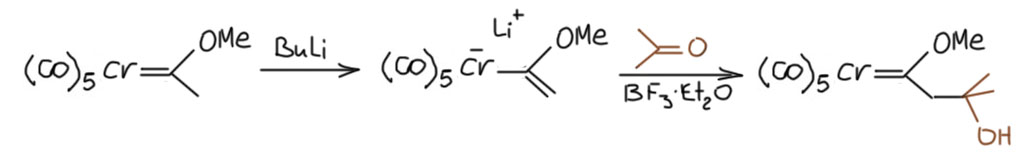
Такие анионы были скоро получены в чистом виде стехиометрическим депротонированием с помощью бутиллития (огромный перебор по pK, но металлоорганики так привыкли к этому реагенту, что используют его везде, где могут. Соли были закристаллизованы и проявили отменную устойчивость в твердом виде (Casey C. P., Anderson, R. L. J. Am. Chem. Soc. 1974, 96, 1230), и реагировали в типичных реакциях для стабилизированных карбанионов.
В этом месте сразу приходит в голову основная реакция всех енолятов и их аналогов – альдольная конденсация. Но это окзалось не совсем просто и понятно почему – слишком большая кислотность означает и низкую нуклеофильность “енолята”. Мы хорошо знаем эту проблему из обычной органической химии, где приходится использовать всякие ухищрения, чтобы решать проблему обратимости альдольных конденсаций. В химии карбенов Фишера пришлось использовать активацию карбонильного соединения кислотами Льюиса. Реакции проще делать с уже готовыми “енолятами”, например (Wulff W. D., Gilbertson, S. R. J. Am. Chem. Soc. 1985, 107, 503). Типичный альдоль (нарисован продукт после обычного гидролиза). Но при этом и новый карбен Фишера, ведь карбеновый центр после реакции возвращается.
В равновесных условиях тоже иногда получается, используя амин и немного неожиданную кислотную активирующую добавку, триметилхлорсилан, но это неудивительно, кремниевые производные за счет огромного сродства к кислороду часто используют в разных вариантах альдольных конденсаций. В реакции образуются продукты дегидратации альдолей – то есть, по-нашему это такая кротоновая конденсация (Aumann R., Heinen, H. Chem. Ber. 1987, 120, 537).
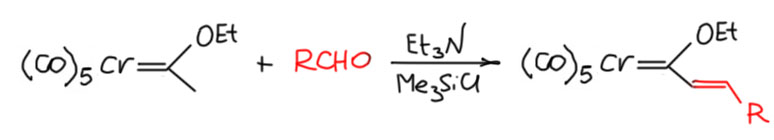
Ещё чаще карбены Фишера используют в реакциях Михаэля, причем и как аналоги енолятов, и как акцепторы Михаэля. Иногда даже и так, и так – и так можно получить двойные карбены Фишера с разными металлами (Macomber D. W., Hung M. H., Madhukar P., Liang M., Rogers
R. D. Organometallics 1991, 10, 737). И даже воспользоваться обычным приёмом в химии карбонильных соединений – при реакции нуклеофила с акцептором Михаэля образуется новый енолят, который можно или сразу погасить донором протонов или поймать ещё одним электрофилом, например субстратом, активным в SN2-замещении. Вот как в этом примере:
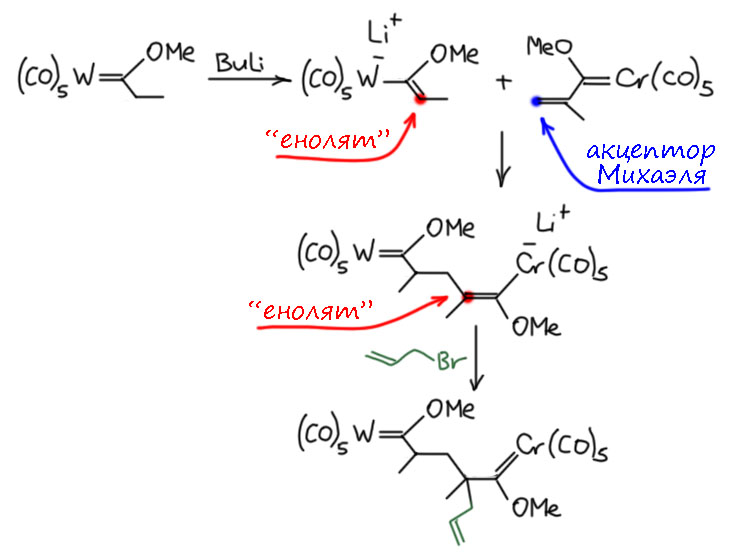
Присоединение по Михаэлю часто применялось в химии карбенов Фишера, так как это гибкая реакция с кучей возможностей для построения сложных структур.
У карбенов Фишера ещё немало других реакций, в основном связанных с электрофильностью, с разнообразными продолжениями и завершениями, очень часто приводящими к циклизациям. Все эти реакции стехиометрические, во многих тем или иным способом из продуктов уходит металл и образуется чисто органический продукт. Это интересная химия, хотя нельзя сказать, что она часто применяется в органическом синтезе, впрочем, есть минимум один метод, бензоаннелирование по Дёцу, который нашёл неплохое применение в синтезе сложных природных молекул. Поскольку там по дороге происходит еще и карбонилирование, мы обсудим этот интересный метод в разделе про оксид углерода.