Комплексы-карбеноиды
В химии карбенов есть понятие “карбеноид” – это нечто, не являющееся карбеном, но ведущее себя в реакциях как карбен. Кстати, в этом смысле мы подвергаем критике популярный утиный тест про то, что если что-то выглядит как утка, плавает как утка, крякает как утка, то это и есть утка. На примере карбеноидов мы видим, что бывают вещи, которые выглядят как карбен (а как выглядит карбен?), реагируют как карбен и дают те же продукты, что карбен, то это может быть не карбен, а карбеноид. То есть и там тоже могла быть не утка, а уткоид. Но дело спасает то, что оргинал утиного теста английский, а в той культуре перед умозаключениями принято добавлять слова likely, very likely, probably и т.д. всегда выражающие гипотетичность любого высказывания, сформулированного на основе наблюдений. Нас это дико раздражает, потому что мы хотим ясности и стопроцентной определённости. И в этом такое фундаментальное различие культур. Мы хотим определённости и поэтому верим в любую ложь, лишь бы она была высказана с авторитетным видом уполномоченным субъектом. А в европейской культуре научного рассуждения любое высказывание подвергается сомнению, которе никогда нельзя устранить полностью, можно только добыть еще некоторое количество доводов в пользу высказывания. Поэтому они смело применяют утиный тест, понимая, что утковидный объект всё же имеет шанс не оказаться уткой, и никакой трагедии в этом нет, мир не перевернётся. А нечто, ведущее себя как заправский карбен – не оказаться карбеном. А оказаться чем-нибудь даже намного более полезным.
У карбенов (настоящих) много типичных реакций – это и внедрение по С-Н связям, и реакция с гетероатомами тоже с внедрением по связи Х-Н или образованием илидов, но почти каждый спрошенный скорее всго скажет, что главнейшая и самая характерная реакция – это циклопропанирование. Даже ещё точнее – согласованное стереоспецифическое циклопропанирование. А когда мы от карбенов переходим к карбеноидам, циклопропанирвоание точно становится главнейшей реакцией, птому что многие циклопропаноиды не способны проявлять себя во всём спектре реакций настоящих карбенов. Все мы знаем реактив Симмонса-Смита на основе цинкорганики. Есть ещё много литиорганических карбеноидов, это целая отдельная наука Это полезные и широко применяемые реагенты, но имеющие общий недостаток всей активной непереходной металлоорганики – они слишком активны и несовместимы с кислыми водородами и многими функциональными группами.
Но есть и карбеноиды на основе производных переходных металлов. И здесь есть небольшая путаница, связанная с тем, что такие карбеноиды в основном идут из одной единственной реакции – разложения диазосоединений. И, по крайней мере, вначале эту реакцию считали источником настоящих карбенов, хотя уже самые ранние исследователи этой интереснейшей реакции (про неё подробнее я как-нибудь расскажу на органическом сайте) отмечали, что карбен, полученный разложением в присутствии какой-нибудь соли (начинали в основном с меди) и карбен из теримического или фотохимического разложения ведут себя по-разному, первые всегда селективнее, и почти не хулиганят, приставая ко всем без разбору слабым связям.
Способность разных солей и соединений переходных металлов (металлов побочных подгрупп) разлагать диазосоединения была обнаружена в начале 20-го века Людвигом Вольфом, открывшим первую реакцию, в основе которой превращение карбена, генерированного из диазосоединения. В перегруппировке Вольфа работают разные переходные металлы, но стандартом стали производные серебра. Точный механизм этой реакции до сих пор не установлен, но представление о том, что в ней участвую комплексы карбенов, является основным. Впрочем, не всё так просто. Исторически первым исследователем, заметившим хитрую особенность реакций разложения диазосоединений в присутствии солей переходных металлов был английский химик Питер Йейтс, в своей диссертации в 1952 году, над которой он работал в Йеле, показал, что те же диазокетоны при разложении в присутствии производных меди не дают никаких продуктов перегруппировки Вольфа, но дают тем не менее продукты, образования которых можно ожидать от карбена. А если мы считаем, что в перегруппировке Вольфа всегда участвует синглетный карбен, даже возможно не успевший толком образоваться, то этот результат и говорит, что что-то там не так, и что металл как-то на результат влияет. Впрочем, в 1952 году Йёйтс даже слова карбен ещё не знает – он говорит про некий двухвалентный радикал, который предполагался интермедиатом в перегруппировке Вольфа. Вообще, хотя это очень важная работа в истории этого вопроса, к ней, конечно, есть и пара претензий. Первая и самая главная – Йейтс выбрал не очень удачные реакции, в первую очередь реакции со спиртами, а про эти реакции, когда с карбенами более-менее разобрались, узнали, что они имеют невероятно высокие скорости и практически вообще не имеют барьеров активации, и что в перегруппировках Вольфа они и так часто успешно конкурируют с перегруппировкой. Ну и можно сказать, что слово карбен уже появилось в 1951 году в работе основоположника этой химии Уильяма Дёринга. Но, безусловно невозможно требовать, чтобы новые идеи так быстро овладевали исследователями. Про Дёринга и начала химии карбенов я расскажу как-нибудь отдельно, а здесь пока отметим, что начиная с 1950-х возникает и сама химия карбенов, и представление о том, что одни из главных предшественников карбено, диазасоединения, могут разлагаться не только термически и фотохимически, но и в присутствии переходных металлов и их соединений.
Дальше некоторое время просто пробуют разные комплексы. Приоритет долго продолжают отдавать двухвалентной меди. Одна из задач – сделать селективным циклопропанирование. В этом месте можно было бы удивиться – а разве циклопропанирование это не основная реакция карбенов, что за проблемы. Это удивление в значительной степени идёт от впечатления, полученного на 3-м курсе, где даже в практикуме делают циклопропанирование и никаких особых проблем не испытывают. Увы, это заблуждение. В практикуме используют всегда один и тот же карбен – дихлоркарбен, генерируемый действием оснований на хлороформ. Но это очень особенный карбен. Он, с одной стороны, стабилизирован, всегда синглетен и не имеет проблем с равновесием синглет-триплет. С другой стороны сильно электрофилен и обладает высокой реакционной способностью и селективностью именно к двойным связям. Поэтому он такой удобный. Увы, таких карбенов немного, в основном это такие же дигалогенкарбены. Большинство же карбенов вовсе не такие благообразные. Реакции большинства карбенов с олефинами весьма капризны и часто неселективны. Проблема даже не в том, что циклопропан будет не той конфигурации – синглетные карбены всегда стереоспецифичны, а в том, что циклопропан не основной продукт таких реакций, и значительная часть карбена пойдет на внедрение по CH-связям. Поэтому циклопропанирование лучше делать не карбенами, а или карбеноидами или металлокомплексами, а последние это как правило и есть то, что образуется при катализе производными переходных металлов.
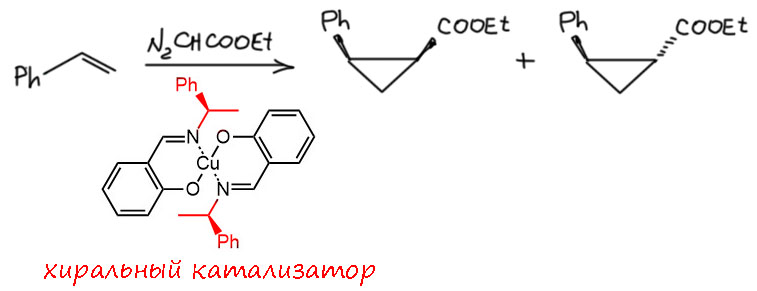
В 1960-е начинается охота за энантиоселективностью. Исследователи ищут реакции, которые можно попробовать сделать энантиоселективными, и пытаются получить хоть небольшой перенос хиральности с самых разных источников. Циклопропанирование кажется отличной моделью для энантиоселективного варианта. Пара заместителей на разных атомах цикла уже дает асимметрию, выбор субстратов поэтому велик и позволяет брать самые простые исзодные. Первая работа принадлежит мощному японскому химику Хитоси Нодзаки, в лаборатории которого начинал свою карьеру будущий нобелевский лауреат как раз по энантиоселективному катализу Рёдзи Ноёри. Первая работа ( Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1966, 7, 5239) хорошо показывает, какой путь прошла эта химия от своего начала. Первая попытка – просто комплекс в составе которого есть хиральный лиганд, хиральность создается одним асимметрическим атомом, висящим где-то в стороне от места события, на роль которого нетрудно предложить собственно атом металла. Итак некий комплекс Cu(2+) катализирует реакцию диазоуксусного эфира со стиролом. Реакция показывает низкую диастереоселективность (получается смесь транс и цис в соотношении около 2:1), и каждый из диастереомеров обладает оптической активностью, впрочем оценка оптического выхода дает мизерные 6%.
Интересно как первый шаг, но неубедительно. В начале 1970-х намечается более серьёзный прорыв. Забавная команда из Льежского университета в Бельгии находит сначала ацетат палладия как более эффективный катализатор циклопропанирования (R. Paulissen, A.J. Hubert and Ph. Teyssié, Tetrahedron Lett. 1972, 1465). Забавная потому, что в каждой следующей статье звёздочка основного автора стоит на другом члене команды, то есть рохоже, что они там жребий кидали каждый раз, кому звездочка достанется. Но именно эта команда делает одно из главных открытий в этой химии – от палладия они переходят к родию, и перебрав несколько комплексов родия они выходят на с виду очень простые соединения – ацетат Rh(2+) и другие соли карбоновых кислот. Именно эти производные оказываются самыми эффективными катализаторами циклопропанирования (Hubert A. J., Noels A. F., Anciaux A. J., Teyssié, P. Synthesis 1976, 600).
Карбоксилаты родия – главные игроки в каталитическом циклопропанировании
Итак, ацетат родия оказался отличным катализатором. Что же это такое, как устроено, почему работает.
Это очень интересная штука и сама по себе, и как типичный представитель большого класса комплексов и даже не только переходных металлов. Во-первых, это димер. Формулу комплекса так всегда и пишут [Rh2(OCOR)4]. Родий в нём имеет степень окисления 2+, то есть нечетное число электронов d7. Именно поэтому это и димер, неспаренные электроны спариваются, так же как рекомбинируют обычные радикалы, хотя тут не всё так просто, но подробное обсуждение отложим до того момента, когда я созрею включить в курс связи металл-металл. Почти уже созрел, но отложу ещё на год.
Ацетаты здесь в общем-то типичные бидентатные мостики, на каждом атоме родия дают MX2L2. Итого, если не учитывать димеризацию это 7+8 = 15 электронов. Димеризация добавляет еще один электрон и получается 16 электронов – координационно ненасыщенный комплекс, есть место еще для одного лиганда. В ацетате родия это просто вода – это дигидрат, и получается что это 18-электронный комплекс, но со слабым лигандом, который легко обменяется на что-то более существенное. Каждый атом родия имеет октаэдрическое окружение, но это свойственно элементам этой группы.
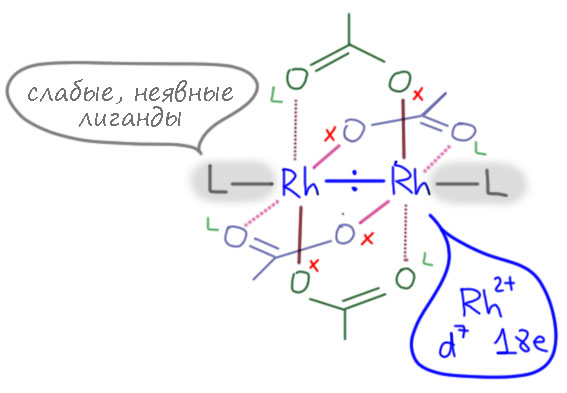
Когда мы на это смотрим, особенно в таком движении, не можем не подумать, что эта штука на что-то похожа. По-моему, на турникет. Видимо, потому что у нас их много. Мы любим турникеты, любим протискиваться через эти уютные устройства, особенно интересно это делать, если у вас в руках чемодан или лыжи, а лучше и то, и другое. Но в тех краях, где придумали название для этого типа структуры, турникеты, видимо, не так популярны, и им пришла в голову другая аналогия – paddlewheel. По-русски, это увы, одним словом не переведешь. Это либо гребное колесо у таких старых пароходиков, как в книжке про Тома Сойера и Гекльбери Финна, ходивших по Миссисипи. Либо, и современному человеку это визуально ближе, это тоже гребное колесо на таких курортных водных велосипедах. Отличная метафора на самом деле.
С тех пор комплексы с мостиковыми лигандами типа paddlewheel (кстати, по идее этим словом нужно бы называть сами комплексы, потому что именно комплекс в сборе напоминает гребное колесо, а лиганды играют роль гребных лопаток, paddles, но не менее часто так называют именно лиганды – paddlewheel ligands) стали весьма популярны в координационной химии. Одно их их потрясающих свойств – высокая устойчивость таких конструкций, они во многих реакциях ведет себя как единое целое и поволяют создавать весьма затейливые молекулярные архитектуры. Но даже сами по себе эти комплексы обычно сохраняются в реакциях. Это интересное свойство, необычное. Мы ведь уже не раз имели дело с димерами, особенно это характерно для комплексов палладия и никеля, но здесь димер – временная конструкция, без связи металл-металл, димер расщепляется в равновесии в растворе и димеризация играет роль координационно лабильного лигандного окружения.
Но здесь все не так. Именно димер работает в реакциях.
Карбеновые комплексы
Одна из удивительных особенностей родиевого катализа в реакциях диазасоединений состоит в том, что а) никто не сомневается в том, что ключевым интермедиатом является карбеновый комплекс родия; б) при этом никто этого комплекса не видел, потому что по всем оценками эти комплексы обладают колоссальной реакционной способностью и время жизни этих комплексов очень мало, что не позволяет как-то их поймать и охарактеризовать. Поэтому некоторые исследователи даже склонялись к согласованным механизмам реакций, в которых такие комплексы рассматривались скорее как переходное состояние в реакциях с субстратами. Но одно исследование всё же есть, в котором такой комплекс был пойман и охарактеризован, а его реакции наблюдались “в прямом эфире”. Для этого пришлось пойти на некоторый компромисс. Дело в том, что большинство диазосоединений для этих реакций образуются за счёт очень удобной реакции диазопереноса из енолизуемых карбонильных соединений, а это значит, что практически всегда в таких карбенах есть одна или даже две акцепторные группы. А акцепторные группы увеличивают электрофильность карбенового углерода, делая комплекс все менее и менее устойчивым (не в термодинамическом смысле, о чём мы ничего толком не знаем, а в смысле реакционной способности – если частица реагирует с максимально возможной скоростью со всем, что может найти рядом, уже неважно, какая у неё энтальпия образования. На очевидный вопрос – а что будет, если в реакционную смесь не добавлять ничего, с чем можно реагировать – следует очевидный ответ, что бешеная собака кусает собственный хвост, и что в таких больших молекулах всегда есть что-то, за что можно зацепиться, и тогда просто происходит совершенно неконтролируемое разложение с образованием кучи непонятных продуктов. И вот, чтобы получить хоть какой-то шанс поймать родиевый карбен, взяли заготовку так называемого пуш-пульного типа (push-pull, тяни-толкай, точнее толкай-тяни – распространённый термин для обозначения ситуаций, когда на каком-то центре нестабильности, радикале, карбене или чём-то ещё, имеющем амбивалентный характер делокализации, висят одновременно и донорный и акцепторный заместители) (Kornecki K. P., Briones J. F., Boyarskikh Y., Fullilove F., Autschbach J., Schrote K. E., Lancaster K. M., Davies H. M. L., Berry J. F. Science 2013, 342, 351). Статья опубликована в журнале Science – это лишний раз говорит о том, насколько эта химия выглядела как прорывная и гламурная, журнал этот в мире естественных наук – вершина гламура, чтобы там что-то опубликовать нужно убедить редакторов, что это и есть самый наипереднейший край науки, и что люди будут с утра очередь занимать в киоски, чтобы урвать номер журнала с этой статьёй, и неделю больше ни о чём говорить не будут и в гламурных гостиных, и в портовых распивочных. ОК, не спорим, пусть будет так, ведь химия родиевых карбенов действительно интересна и довольно парадоксальна.
Взяли диазокетон, содержащий донорную п-метоксифенильную группу. И попробовали много разных карбоксилатов родия, пытаясь поймать интермедиат. С одним удалось, весьма громоздким трифенилацетатом, вероятно помогла как раз стерика, немного затрудняющая доступ других молекул к карбеновому центру. Реакция с диазо-производным метилового эфира п-метоксифенилуксусной кислоты действительно дала нечто в растворе, что удалось обнаружить с помощью разных видов спектроскопии, в том числе даже ЯМР: в углеродном спектре обнаружился очень слабопольный сигнал карбенового углерода аж при 240 м.д., что выдаёт его острую электронодефицитность. Интересно, что этот сигнал в углеродном спектре расщеплён в дублет на одном из изотопов родия, у которого самый обычный для ЯМР спин в 1/2.
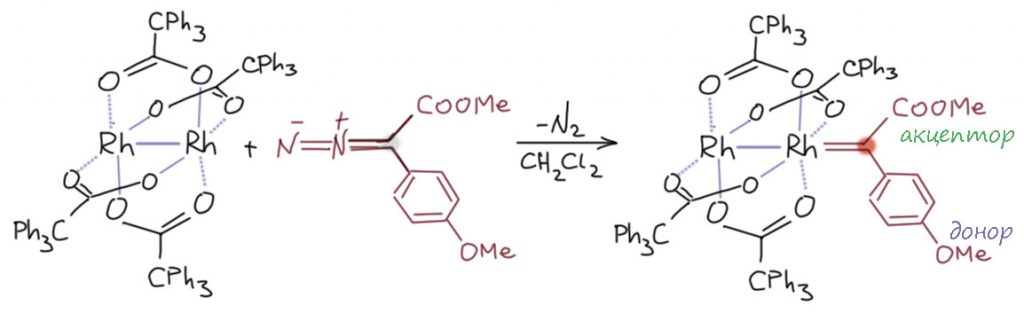
Комплекс этот живёт недолго – время полужизни всего 10 секунд (вспомните кинетику, если забыли, что такое время полужизни, halflife time), но это колоссально много по сравнению и со свободными карбенами (стабильные карбены не в счёт, это особый тип), и всеми другими родиевыми карбеноидами, которые до этой работы не удалось поймать ни разу, да и после этой работы, а уже почти 10 лет прошло, кажется, тоже.
Реакции у этого карбена самые обычные – циклопропанирование, внедрение по связям CH, присоединение соединений с гетероатомами, а если вообще ничего нет, то с самим собой. Их можно делать или в каталитическом варианте, или сначала сгенерировав комплекс. Выходы в последнем случае безусловно ниже, так как мы не умеем делать реакции с соединением, которое живет 10 секунд, конечно, оно частично успееет разложиться. В реакциях внедрения по СН-связям карбеноид селективен, например, в ТГФ внедряется только в всязь на альфа-углероде.
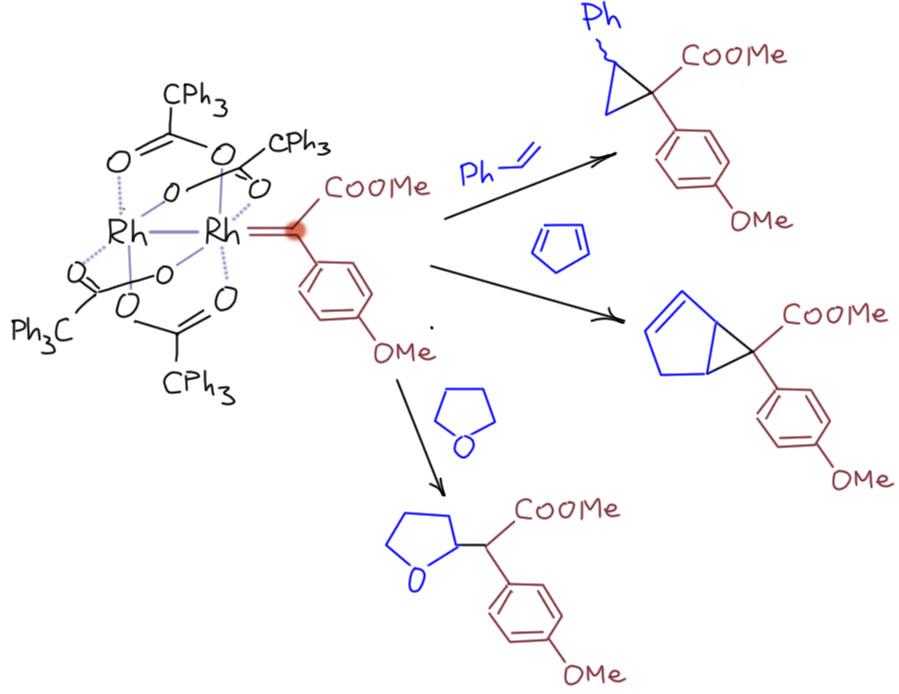
А зачем связь родий-родий и вообще вся эта конструкция??
Мономерные комплексы родия, даже такие простые как хлорид, тоже отлично разлагают диазосоединения и вызывают каталитическое циклопропанирование и другие реакции. Но эти димеры типа “гребное колесо” во-первых, работают намного эффективнее, гораздо выше и TON и TOF, а кроме того, они селективнее и могут быть легко использованы для диастереоселективного и энантиоселективного циклопропанирования. Поэтому эти красивые штуки стали стандартом этой химии, предметом изучения и восхищения – вообще это большое удовольствие изучать реагент, который и работает хорошо, и весь сам из себя изумительно прекрасен. В этот момент учёный совершенно подсознательно понимает, что нашёл одну из тайн Природы (или Творения, кому как нравится), а не просто вымучивает очередную сттатейку для гранта или диссера.
Итак, почему димерный реагент с связью металл-металл круче мономерного? На самом деле мы здесь снова погружаемся в ту же историю, что уже видели на карбенах Фишера – тонкую настройку эффекта back-donation. Этот эффект нужен для стабилизации карбенового лиганда, потому что просто висеть на координационной связи можно только если в самом карбене сильна собственная стабилизация, как в NHC и их аналогах – стабильных нуклеофильных карбенах. А когда карбен слабо стабилизированный или вовсе дестабилизированный, с акцепторными заметителями, некоторый back-donation требуется, иначе эта штука сорвётся с цепи и начнёт крушить всё подряд, не разбирая реакционных центров. Получается, что в химии таких карбенов тонкая настройка back-donation – это ключ к большим достижениям. Мало – будет слишком неустойчив и необуздан, неселективен. Много – будет ленив и вообще ничего не захочет. Родий сам по себе – неплохой донор back-donation в валентных состояниях с конфигурацией d6, то есть Rh(3+) – это в низкоспиновом состоянии диамагнитные комплексы. Неплохой, но недостаточно хороший, недолёт. Когда мы создаём связь Rh-Rh в валентном состоянии Rh(+2), мы спариваем неспаренный электрон, и фактически получаем конфигурацию d8, а это уже даже из общих соображений должно сопровождаться усилением back-donation.
При желании можно посмотреть, как это представляют на уровне молекулярных орбиталей. В этом случае рассматривают трёхатомный фрагмент – два атома родия и карбеновый центр, и приходят к модели, которая очень часто всплывает в разных местах химии, прежде всего в тех, что связаны с гипервалентными соединениями – модели трёхцентровой связи, хотя здесь она в некотором смысле вывернута наизнанку. Эта теория развита в нескольких работах, самая ранняя принадлежит Эйити Накамуре с сотрудниками ( E. Nakamura, N. Yoshikai, M. Yamanaka, J. Am.Chem. Soc. 2002, 124, 7181), хотя в то время расчеты переходных металлов давались ещё тяжело. Позже модель развил Джон Берри, много занимавшийся химией диметальных катализаторов не только карбоновой химии (J. F. Berry, Dalton Trans. 2012, 41, 700).
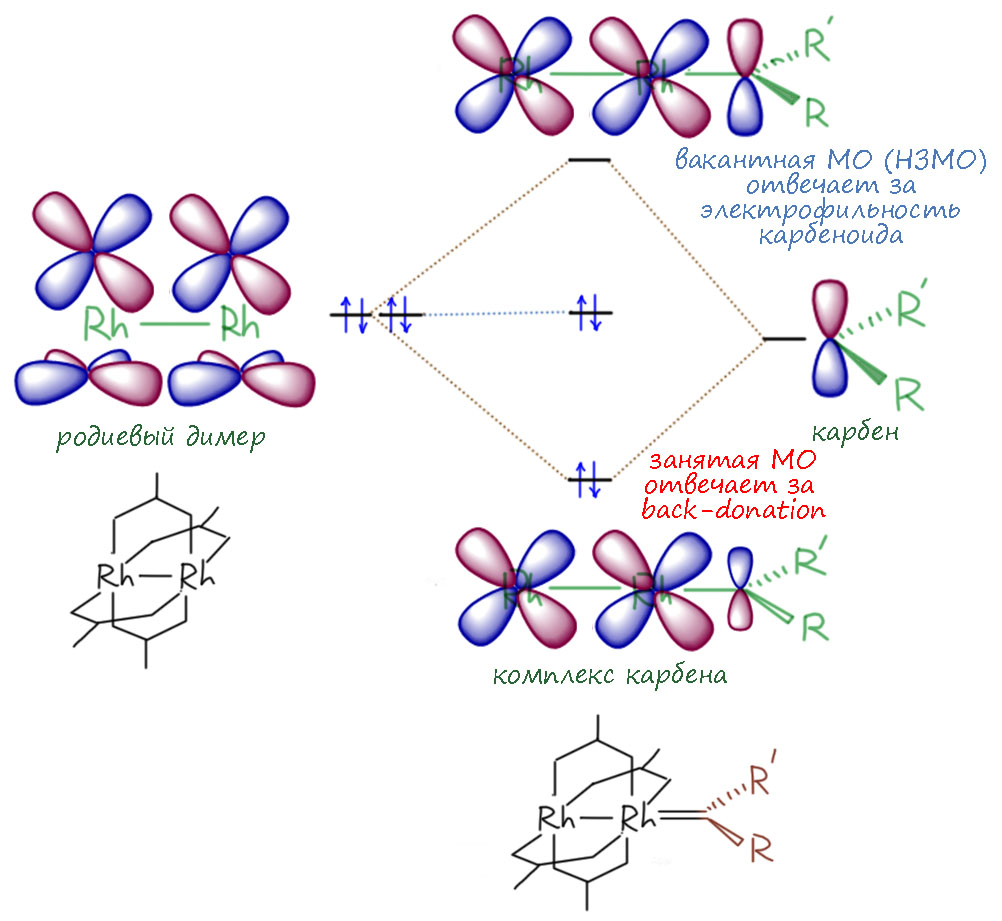
Как идёт реакция циклопропанирования?
Теперь мы более-менее знаем, как устроен карбеновый комплекс родия. Важно то, что на карбеновом углероде остается вакантная p-орбиталь, которую мы или рассматриваем как таковую, с учетом некоторой стабилизации за счёт умеренного эффекта back-donation, или мы используем представление о трёхцентровой связи, и тогда мы видим НСМО с очень высоким весом той же самой p-орбитали и антисвязывающей комбинацией двух d-орбиталей атомов родия. Особенно этот последний подход говорит нам, что мы имеем дело с центром огромной электрофильности, в этой науке часто всплывает броское слово “суперэлектрофильный” из работ Джона Берри, но это мы оставим на совести любителей кликбейтной рекламы. В отличие от термина супернуклеофильность, которым мы пользуемся и видим в этом смысл в химии нуклеофильного замещения, здесь совершенно явная спекуляция, ведь карбены сами по себе и так обладают колоссальной электрофильностью, практически не имеющей равных в мире реакционноспособных интермедиатов. Связь с родием всё же скорее немного притушивает реакционную способность – это точно можно сравнить с укрощением строптивой – ведь в этой пьесе Шекспира речь идёт не о том, чтобы сломить сопротивление и сделать Катарину покорной и безвольной в стиле “жена да убоится мужа своего” и “церковь, кухня, дети” как любят некоторые убогие реликты домостроя – цель Петруччо скорее договориться о разумном следовании некоторым принятым нормам жизни, но сохранить при этом яркую индивидуальность избранницы – собственно у него и выбора другого нет, потому что иначе его просто пошлют вслед за известным кораблём и сделка не состоится. Вот и здесь такая картина – связь карбенового лиганда с родием оставляет эту орбиталь для электрофильных взаимодействий, и реакционная способность остается огромной, при этом реакции приобретают некоторую благообразность: селективность – двойная связь дает циклопропанирование, внедрение происходит только по активным C-H связям, иногда бывают и другие реакции.
Реакцию циклопропанирования много раз моделировали путём квантово-химических расчетов разной стпени сложности. Главная идея, которая при этом вырисовывается – медленной стадией неожиданно является образование карбенового комплекса из диазосоединения, то есть фактически стадия загрузки лиганда на металл. Впрочем а что тут необычного – в реакциях, где первой стадией является окислительное присоединение, в том же кросс-сочетании, первая стадия очень часто является скорость-определяющей. Вот и в этой реакции – что здесь кстати происходит? – сначала диазосоединение, являющееся углеродным нуклеофилом, что видно по одной из граничных структур, просто вступает в реакцию лиганного обмена – неявный лиганд на родии меняется на диазосоединение, но за этим немеделенно следует обычное альфа-элиминирование азота:
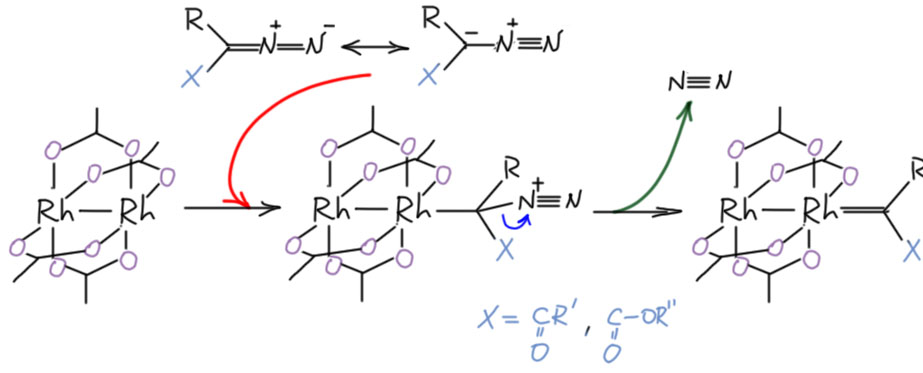
Дальше реакция идёт как электрофильная атака на двойную связь, и это очень похоже на реакцию карбокатиона, но на втором углероде положительный заряд не успевает развится, как туда переползает та пара, которая обслуживала координационную связь карбенового центра с металлом – и сразу получается циклопропан. Поэтому мы можем нарисовать только переходное состояние, где есть формальное разделение зарядов, ведь при развитии процесса свободная орбиталь на карбеновом атоме уходит в дело, back-donation наоборот больше не у дел, и происходит нечто очень похожее на то, что мы рисовали в реакциях карбенов Фишера – временное превращение L-лиганда в X-лиганд, и возникновение формального заряда на металле, иначе мы изменим степень окисления, а нам это ни с какого конца не нужно.
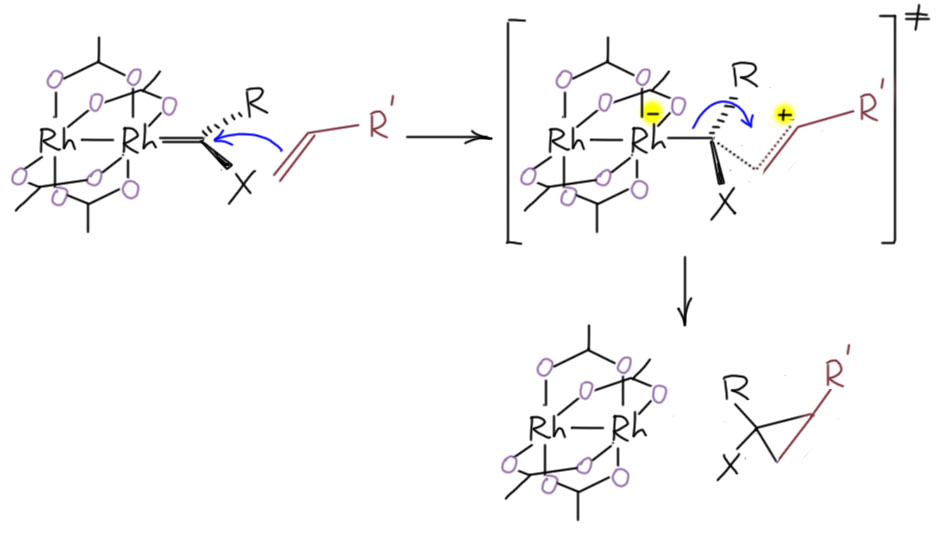
Раньше иногда постулировали промежуточную стадию, когда возникает металлациклобутан, далее подвергающийся восстановительному элиминированию, но большого смысла в этом дополнении нет, да он и предполагает немного иную траекторию приближения олефина.
Стабильность карбеновых комплексов родия
Исследования родиевых карбеноидов и их реакций за тридцать лет помогли найти хорошие катализаторы для стереоселективных реакций, но, как ни странно, гипотеза о том, что в этой реакции образуются карбеновые комплексы не была подтверждена экспериментально выделением и описанием таких комплексов – они оказались чрезвычайно реакционноспособны. Статью в Science двух главных исследователей этой химии, Джона Берри и Хью Дейвиса, в которой был спектроскопически пойман такой комплекс, мы уже рассмотрели. Казалось, что это максимум того, что можно сделать, недаром и статью тиснули в такой гламурнейший журнал. Но эти ученые сами того не желая разбудили настоящего монстра химии переходных металлов, швейцарца Алоиза Фюрстнера из знаменитого института Угля общества Макса Планка в Мюльхайме. Фюрстнер немного подумал, и выстрелил целой серией работ, в которых были впервые получены и сами комплексы, и их структура охарактеризована рентгеноструктурным анализом. И ничего особенного Фюрстнер к уже известной химии не добавил, просто работа была сделана мастерски с точки зрения эксперимента и с точки зрения замысла, и сразу показала, что все невероятные мучения с многолетней ловлей родиевых карбенов была скорее приговором тем, кто этим занимался.
Первая работа (Werlé C., Goddard R., Philipps P., Farès C., Fürstner A. J. Am. Chem. Soc. 2016, 138, 3797) была особенно показательно своей удивительной простотой – надо было просто подобрать правильные карбоксилат и карбен и всё получилось. Чтобы стабилизировать карбен получше, взяли два донорных фенила. Бис(п-метоксифенил)диазометан получен по Штаудингеру окислением гидразона бензофенона оксидом ртути. Это диазосоединение вполне нормально реагирует с типичным олефином, п-метоксистиролом в присутствии 0.5 моль% трифенилацетата родия при -10°С, и с тем же соединением родия образуется вполне стадильный карбеновый комплекс, который можно при небольшом минусе некоторое время хранить в растворе, снимать спектры. Удивительно, но в углеродном спектре сигнал аж при 268 м.д., ещё дальше чем у карбена с заместителями типа донор-акцептор, описанного в той статье в Science. Но это неплохо соотвеnствует тому, что в этом комплексе самое длинное расстояние Rh-C, а это приводит к очень интереcному выводу – если карбеновый центр стабилизирован сопряжением с обычными донорными заметителями, back-donation становится не таким важным и проявляется слабо. А это говорит, что в ряду карбенов на родиевом димере очень важен баланс взаимодействий. Долго возились, но смогли получить структуру при -20°С, она немного разупорядочена, но пригодна. Второй атом родия связывает кислород из метокси-группы, получается полимерная и немного кривая структура. Структура QUZTIQ описана в кратком сообщении (Werlé C., Goddard R., Fürstner A. Angew. Chem., Int. Ed. 2015, 54, 15452). В структуре комплекса очень хорошо видно, как глубоко утоплен карбеновый центр в нагромождении заместителей, висящих на карбоксилате – и это очень хороо соответствует тому, настолько селективна эта родиевая химия к структуре олефина: стерические препятствия просто не дают большинству олефинов кроме терминальных и цис-диамещённых подходить к карбеновому центру.
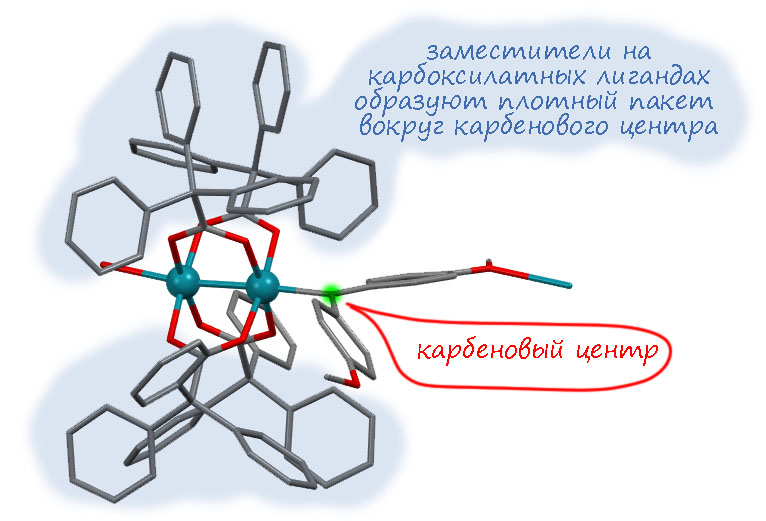
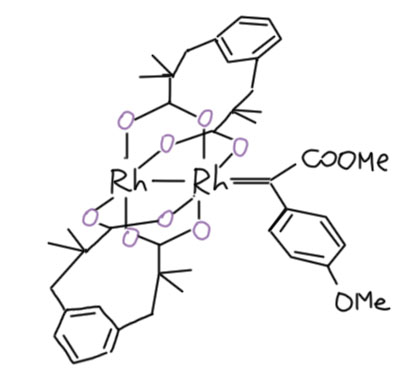
Фюрстнеру и сотрудникам удалось сделать и структуры с донорно-акцепторными карбенами, в том числе тем же, что и был использован в статье в Science, но с немного другим, бидентатным дикарбоксилатом. Связь Rh-C в этом комплексе действительно оказалась короче, а сигнал в углеродном спектре не так слабополен как в предыдущем и неплохо соответствовал тому, чот намерили в Science. Уф, хоть не наврали, гламурщики, отлегло. Структура комплекса ещё раз хорошо показывает, как может и как не может подходить олефин, обеспечивая стереоселективность.
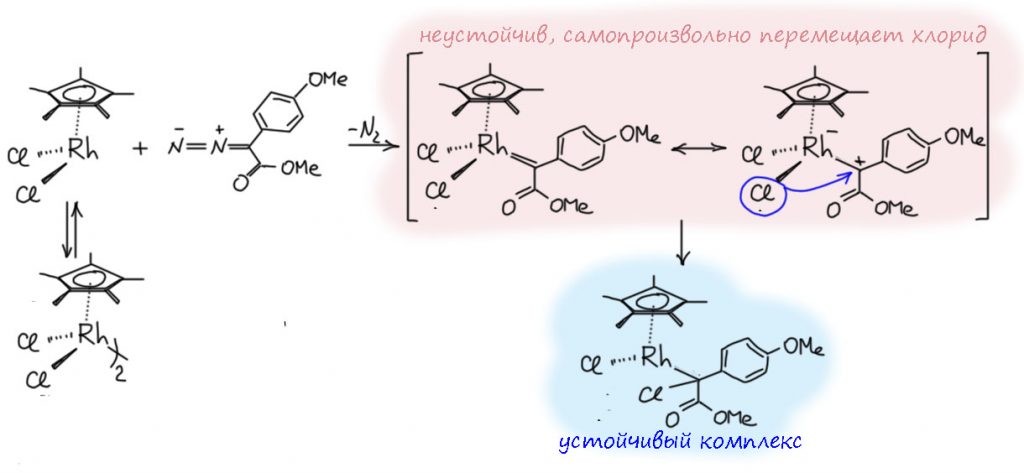
Разбуженный Фюрстнер пошёл и ещё дальше, неявно усомнившись в совсем уже священной доктрине о невероятной уникальности именно диродиевых кластеров для работы с карбеноидами. Как бы ни были интересны гребные колёса, но одноядерные комплексы родия, особенно с циклопентадиенильными лигандами тоже неплохо работают в этих реакциях. Фюрстнеру и сотрудникам удалось получить несколько карбенов с мономерным родием. Оказалось, что один родий даже с более донорным пентаметилциклопентадиенилом (металлоорганики любят этот донорный лиганд и нежно называют “цепе-со-звездой” Cp*) настолько слабо дает back-donation, что эти комплексы лучше показывать не с двойной связью, а в виде структуры с разделением зарядов (напомню, что заряд на металле позволяет не менять степень окисления, ведь карбеновый лигандв этом случае рассматривается как X-лиганд, да ещё и карбокатион). И если на родии хлориды, то происходит самопроизвольное смещение хлорида на карбокатион, а родий избавляется от необходимости иметь формальный отрицательный заряд. Обратите ещё внимание, что хранится исходный комплекс в димерном виде (через галогенидный мостик), и в растворе происходит спонтанная диссоциация с образованием каталитически активного мономера.
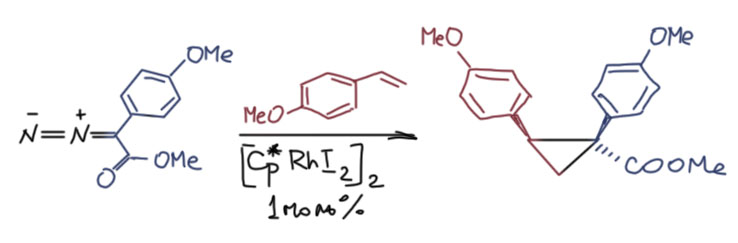
А вот если на родии намного более стабильные бромид или иодид, карбен оказывается устойчив настолько, что удалось сделать рентгены. И в углероде химсдвиг карбенового углерода находится при совсем уж неприличных 314-316 м.д.! А вот расстояние Rh-C короче, чем в диродиевых карбенах, и это говорит о том, что в тех связь действительно возможно более слабая, потому что трехцентровая. Здесь мы имеем ещё один парадокс – стабилизация карбенового лиганда не связана однозначно с эффективностью back-donation: в мономерных родиевых комплексахх связь с карбеновым центром прочнее, но углерод намного более карбокатионный. Кто пустил Фюрстнера в этот огород – скоро ни от одного лозунга, добытого десятилетиями непосильного труда всех остальных не останется и следа и всё придется делать заново!
Такие комплексы отлично катализируют циклопропанирование и внедрение по CH-связям, а также другие интересные реакции. Иодидный комплекс оказывается намного более активным, чем хлоридным, совершенно очевидно потому что карбен устойчивее.
Пока оставиновимся здесь, но я не свожу глаз с работы Фюрстнера и его сотрудников и мы к ним еще вернемся.
Рутений успешно сыграл в ящик
Родиевое циклопропанирование – главный игрок в этой области, но некоторые другие металлы тоже дают неплохие методы, а по цене любой металл выигрывает у родия порядки цены. В главной троице катализа RuRhPd все три металла способны катализировать циклопропанирование диазосоединениями, действуя через образование карбеноидов. Впечатляющие возможности рутения, самого дешёвого из великой троицы, были продемонстрированы японским химиком Хисао Нисиямой и сотрудниками (Nishiyama H., Itoh Y., Matsumoto H., Park S.-B., Itoh K. J. Am. Chem. Soc. 1994, 116, 2223). Эта возможность возникла благодаря открытию одной из самых важных групп хиральных лигандов для катализа – бис(оксазолидинов), обычно сокращаемых как бокс-лиганды (BOX-ligands). Лиганды этого типа фактически открыл – и не заметил этого – швейцарский химик Андреас Пфальц в 1986, когда предложил одно сложное природное соединение в качестве хирального лиганда. Посмотрев на эту молекулу, возбудились сразу несколько учёных, активно искавших новые мотивы для дизайна энантиоселективных катализаторов, а эт были годы, когда эта модная наука дала первые выдающиеся результаты, и немедленно привлекла к себе толпы жаждавших быстрых успехов исследователей, особенно много таких тогда было в Японии. Исследователи сообразили, что это просто великолепная идея, потому что оксазолидины элементарно получаются из аминоспиртов и производных карбоновых кислот (нитрилов, сложных эфиров) – доступных нитрилов и эфиров море, а хиральные аминоспирты просто валяются под ногами – это продукты восстановления аминокислот алюмогидридом лития. Первой такой работой кажется было исследование группы Сатору Масамунэ, японца, всю жизнь, как Негиси, работавшего в США в МТИ. Если два одинаковых по конфигурации аминоспирта повесить на любой остаток дикарбоновой кислоты, получаем лакомое – C2-симметричный хиральный хелатор, а именно этот структурный принцип произвел революцию в асимметрическом катализе (нобель 2001 года – Ноулз, Ноёри, Шарплесс – весь про это, у каждого из этой троицы главное достижение было связано с С2-симметричными лигандами). Масамунэ был первым, поэтому взял самое простое – малоновый эфир, и сделал на его основе ряд бокс-лигандов (Lowenthal R. E., Abiko A., Masamune S. Tetrahedron Lett. 1990, 31, 6005).
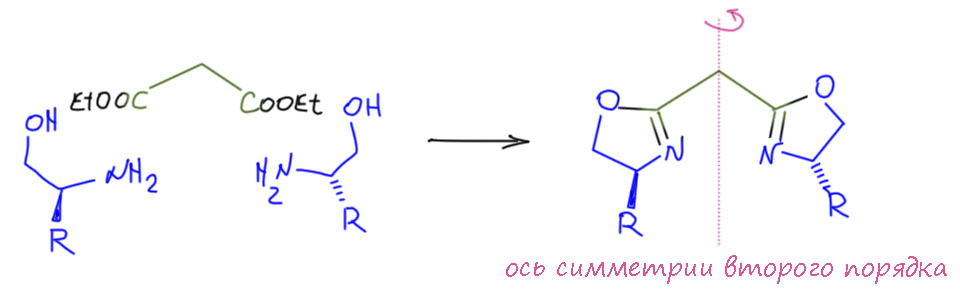
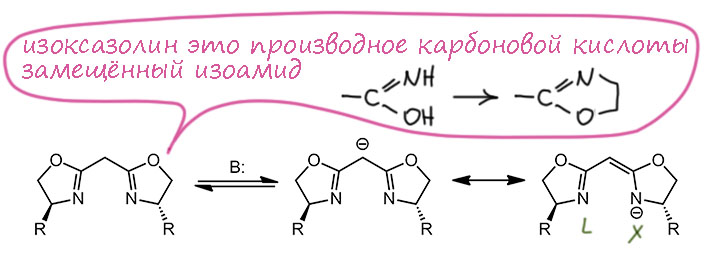
С этими лигандами получили медные комплексы. Поскольку имидазолин – это такое производное карбоновой кислоты, то и весь лиганд ведет себя как производное малоновой кислоты, то есть, например, легко даёт мезомерно делокализованный моноанион, который и работает как XL-хелатор:
И вот из таких лигандов с медью получаются очевидные бис-хелаты и их и используют в реакции циклопропанирования. Оценим путь, который прошла каталитическая химия за последние несколько десятилетий. Работа Масамунэ опубликована в 1990, но она ещё очень простая, в ней не пытаются как-то спроектировать каталитически активный комплекс. Конечно, это зависит от области, потому что в метатезисе к этому времени Шрок и другие уже очень далеко зашли именно в целенаправленном дизайне каталитически активных комплексов, но здесь просто навесили лиганды, сколько навесилось – и в дело. Никто не задает вопросы – а как этот комплекс быдет работать, а есть ли у металла координационные возможности, а не нужно ли расчистить место, снабдив его лабильным лигандом и т.п. Комплексы сработали, получилась и диастереоселективность, хоть и невыдающаяся, и у каждого диастереомера ещё и энантиоселективность, и у самого лучшего лиганда – с трет-бутильной группой, для транс-диастереомера даже вполне высокая.
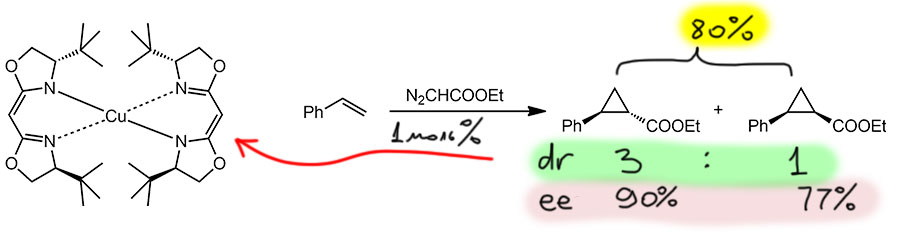
Получилась простая, но очень важная работа, которая показала потенциал нового типа лигандов. Ведь это та же самая медь, что была у Нодзаки в самом начале энантиоселективного циклопропанирования, а насколько лучше результаты! И лиганды не сложнее, а даже проще и возможностей развития намного больше. Это очень важно – лиганды с перспективой должны быть эффективны, но при этом максимально просты и доступны. Тогда это задел для большого развития.
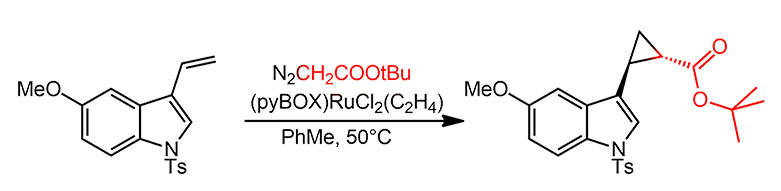
Всего несколько лет спустя исследование получило продожение. 1990-е, как мы уже много раз убеждались, это десятилетие крупнейших достижений в катализе комплексами переходных металлов. К этому времени накопилась критическая масса данных, и пошло более глубокое осмысление, в свою очередь реализовавшееся в решении многих проблем, появлению нновых методов и реакций. Исследователи неуклонно смещаются к катализаторам с определенной координационной сферой, к более точному управлению реакционной способностью и селективностью с помощью специально подобранных анциллярных лигандов. Вот и здесь, от случайного комплекса меди, Нисияма с сотрудниками переходят к комплексу определенной структуры. Для этого потребовалось сделать бокс-лиганды с более жесткой структурой. Нисияма повесил боксы на пиридин, получив тридентатный L3-лиганд. Сейчас бы мы назвали это пинцером, да это слово существовало и тогда, но ещё не было раскручено, и Нисияма не обратил на это внимания. Но факт в том, что такой лиганд (его называли пайбокс – pyBOX) жёстче сидит на металле и точнее определяет геометрию комплексов. Собрали с этим лигандом комплекс Ru(2+) и нашли, что если свободное место заткнуть этиленом, комплекс можно хранить, что впрочем необязательно, так как очень большая константа связывания pyBOX делает возможным класть в реакционную смесь самый популярный рутениевый предкатализатор – димерный комплекс с цимолом – и лиганд. Получилась и диастереоселективность получше, и у каждого диастереомера энантиоселективность.
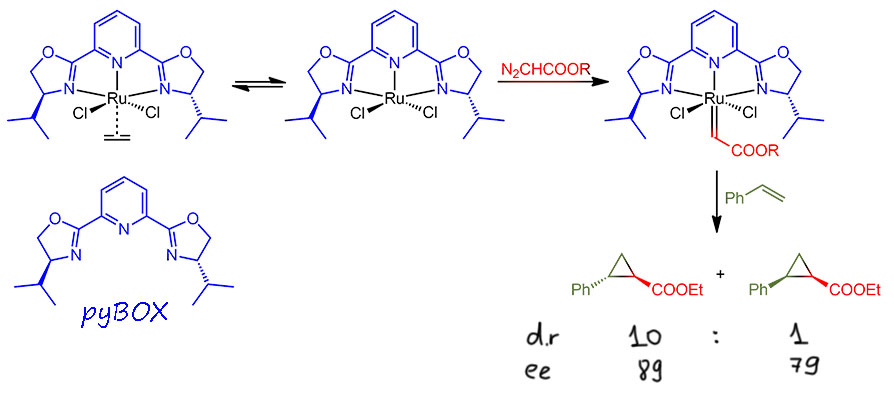
Этот метод, циклопропанирование по Нисияме, вполне прижился в синтезе, и его стали использовать довольно часто. Особенно высокая диастереоселективность получается, если использовать более объёмистый трет-бутил диазоацетат. Так, исследователи из британской фармацевтической компании Бристоль Майерс-Сквибб применили этот метод для синтеза аналогов серотонина с выходом на килограммовые количества продукта (Marcin L. R., Denhart D. J., Mattson R. J. Org. Lett. 2005, 7, 2651). Соотношение транс-цис в случае трет-бутилового эфира получается около 20:1, то есть можно считать, что получается чистый транс-диастереомер