
Карбены и комплексы карбенов
Мы хорошо (так ли это?) знаем, что такое карбены, из обычной органической химии. Там это полезный реакционноспособный интермедиат, участвующий во многих важных реакциях, но точно уступающий по влиянию и встречаемости карбокатионам, карбанионам, радикалам. В химии переходных металлов карбены играют очень важную роль. Долгое время, впрочем, это была забавная экзотика, которой занимались немногие. С открытием и развитием метатезиса, карбены переместились на передний план и стали интересовать многих. В последние 10-15 лет карбены в химии переходных металлов стали, пожалуй, самым модным типом структуры. Их обнаруживают везде, даже там, где никто этого и не ожидал. Карбеновые лиганды наступают с неотвратимостью коронавируса, но никто не объявляет карантин, ни даже самоизоляцию, поэтому они неотвратимо и очень скоро захватят всю химию, мы перепишем все механизмы и каталитические циклы, включив в них карбены.
Пока что мы в начально стадии этой эпидемии (может, эпихимии?). Они наступают. Разберёмся немного, чтобы встретить с достоинством обречённых эти назойливые частицы.
Стабилизированные карбены
Карбен – это половинка олефина по двойной связи точно так же, как радикал это половинка простой связи, а карбин – половинка тройной. Поэтому карбен можно представить себе как sp2-гибридный углерод, на котором висит два заместителя – поэтому это двухвалентный углерод – и две орбитали, занятая гибридная в той же плоскости, где два заместителя, и пустая чистая p-орбиталь, перпендикулярная плоскости sp2-гибридного атома. Это не значит, что все карбены таковы, есть и дургие структуры, но большинство карбенов, интересующих органического химика, таковы, а до других нам пока дела нет. Такие карбены называются синглетными. Исходный карбен (как жаль, что в русском языке нет нормального аналога слова parent – по-английски сказали бы коротко the parent carbene, а как это по-русски? – только чтобы не коряво и без ненужных аллюзий к монументу на Мамаевом кургане) в основном состоянии триплетен. Замещённые карбены в основном синглетны с вакантной p-орбиталью. Поэтому карбен можно представить себе, как карбанион и карбокатион в одном флаконе и даже на одном атоме углерода. Если водороды заместить на другие группы получим замещённые карбены. Мы хорошо знаем, что карбокатионы и карбанионы нуждаются в стабилизации и много внимания уделили этому. Карбокатионы стабилизируются донорными заместителями, +М намного лучше, чем +I, и гиперконъюгация не помешает. Карбанионы стабилизируются акцепторными эффектами, и опять -M лучше, чем -I, и гиперконъюгация тоже не мешает (в случае стабилизации карбанионов гиперконъюгацию принято более солидно называть σ-π*-сопряжением, мы встречались с этой штукой, например, в объяснении, почему так легко депротонируется 1,3-дитиан). Получается что карбен, и карбанион, и карбокатион должен стабилизироваться вообще всем на свете – и донорами, и акцепторами всех типов. Это и так, и не совсем так, потому что эффекты по-разному действуют на две орбитали карбенов. Просто из общих соображений можно понять, что карбену гораздо больше неудобств доставляет не занятая, а свободная орбиталь – она выше по энергии и сильнее дестабилизирует карбен. Не забываем, что углерод всё же неметалл, и лишиться электронов для него неприятнее чем приобрести немного лишних. 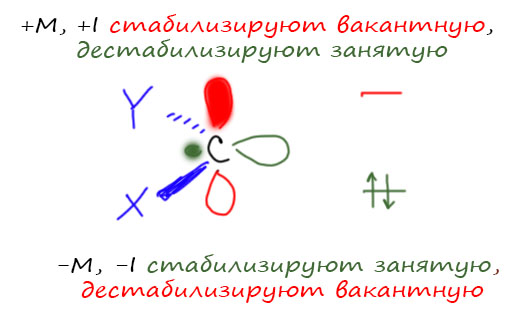
Поэтому мы гораздо чаще увидим карбены с +M-заместителями, чем с акцепторами обоих типов. Акцепторы стабилизируют пару, а ей и так неплохо, но дополнительно дестабилизируют вакантную орбиталь. Получим нечто очень нестабильное и бешено реакционноспособное.
Когда у нас есть +M-заместитель, имеем обычную для химии историю, потому что такие заместители всегда имеют -I-эффект. У галогенов эти два эффекта конкурируют, поэтому получаем хорошо известный из органической химии эффект – дигалогенкарбены легко образуются (значит стабилизированы – работает +М-эффект), но электрофильны – работает -I-эффект.
Если мы повесим на углерод один, а лучше два гораздо более сильных +М-заместителя, а ничего лучше азота с неподелённой парой и придумать невозможно – мы получим мощную стабилизацию вакантной орбитали, которая фактически перестанет быть вакантной. Карбен фактически превратится в карбанион, нуклеофил, основание Бренстеда-Лоури. 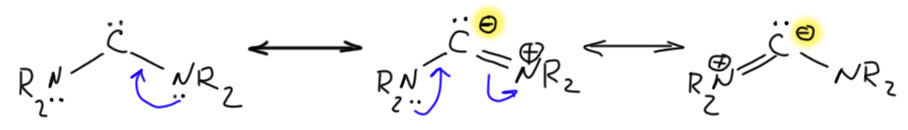
А почему мы называем это карбеном? Потому что самый надёжный формальный признак карбена – двухвалентность углерода, понимаемая как в 19 веке, когда это понятие и было придумано – число ближайших атомов, с которыми можно предположить наличие химической связи. В те времена не было никаких мезомерных эффектов, электронов и прочей дребедени, были только атомы, и некоторое представление, что они как-то связаны друг с другом. Когда мезомерные эффекты появились, мы получили возможность нарисовать граничные структуры, и сказать, что среди них есть карбеновые, а есть, например, как в этом случае – карбанионные.
Одну вещь важно понимать очень хорошо – не существует какого-то определённого свойства или типа реакционной способности, которое выдаёт в частице карбен. Вот карбанион мы распознаём по нукелофильности и основности. А карбокатион по электрофильности и кислотности Льюиса. А карбен может быть и тем, и другим, сразу или по отдельности, а может и не быть. Более хамелеонистой частицы, чем карбен, в химии нет. Поэтому мы их и узнаём то там, то сям, в самых разных ситуациях, и поэтому же до некоторого момента их не распознавали нигде – старая химия была более определённой и любила твёрдо ассоциировать конкретные свойства и типы реакционной способности с конкретными интермедиатами и типами реакционноспосбоных частиц. А новая приобрела свойство видеть всё во всём и везде, отчего её так понравились карбены и она тут же стала их узнавать, как добрых приятелей.
Прототипом карбена с двумя +М-заместителями является такая фундаментально простая частица, как цианид-ион. Цианид-ион – это карбанион, нуклеофил (довольно слабый) и основание (тоже не очень сильное, приблизительно равное обычным алифатическим аминам). Но мы знаем кроме цианида ещё изоцианид. Но отдельного изоцианид-иона не бывает. Цианид и изоцианид это одно и то же, когда мы говорим про анионы. Это просто две граничные структуры одной и той же частицы, которую обычно принято называть цианидом, но можно с тем же успехом называть изоцианидом. Изоцианид – карбеновая форма, цианид – карбанионная. И ещё раз взываю не путать граничные структуры и мезомерию с настоящим равновесием. К какой форме ближе реальный цианид-ион? К цианидной, карбанионной, конечно. В карбеновой, изоцианидной форме рядом находятся вакантная p-орбиталь на углероде и заполненная на азоте – понятно, что такая полностью поляризованная форма не очень выгодна, хотя в ней электроны и смещены на более электроотрицательный атом. Но некоторый вклад в структуру цианида карбеновая форма даёт.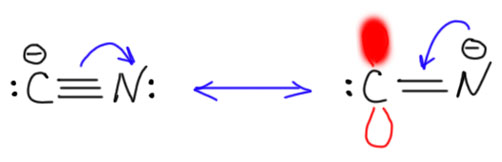
Проявляется ли как-нибудь карбеновая природа цианид-иона? Как мы уже выяснили, трудно определить какое-то одно свойство или реакцию, свойственную всем карбенам. Один очень простой тест есть – реакция с электрофильным кислородом или серой даёт карбонильную или тиокарбонильную группу. Электрофильный кислород – это например, перекиси или гипохлорит. Электрофильная сера – просто сера. И да, и та, и та реакции есть, и в простой химии, и в биохимии. Образуется цианат и тиоцианат-ионы. Так, например, обезвреживают цианиды. 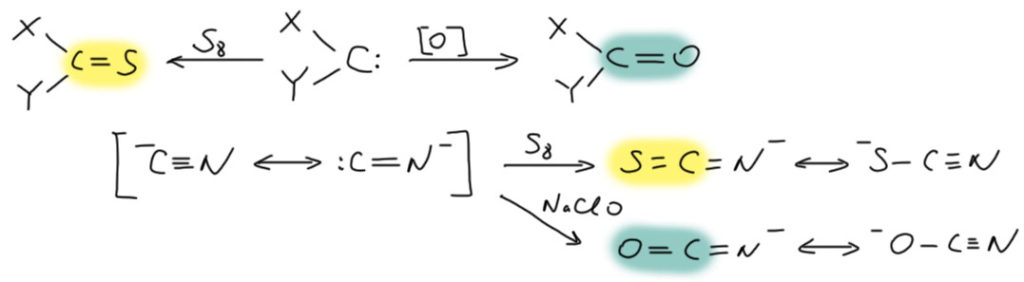
Карбеновую изоцианидную форму легко зафиксировать, превратив анион в изоцианиды – стабильные и фантастически зловонные вещества. Не будем здесь вспоминать всю эту химию амбидентного нуклеофила цианид-иона, просто признаем, что как-то получить изоцианиды можно. Изоцианиды – типичные стабильные карбены. Граничные структуры у изоцианида имеют такую же природу, но несложно заметить, что в этом случае карбеновая структура выглядит не хуже карбанионной с разделением зарядов, хотя именно вторая структура отвечает за нуклеофильность изоцианидов, например, в реакциях Уго и Пассерини, в которых изоцианид присоединяется к карбонильным группам. 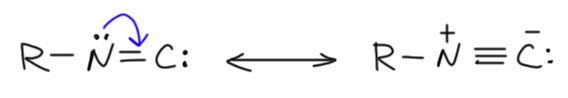
Цианид-ион очень хорошо моделирует карбены с двумя сильными донорами типа амино-групп. Это видно даже просто сравнением структур.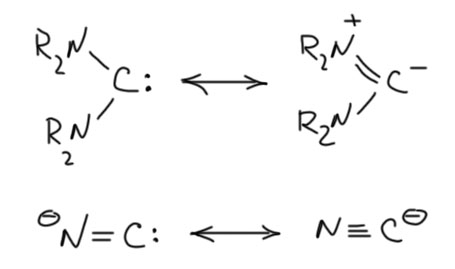
Карбеновая форма с двухвалентным углеродом – две связи к азоту. Карбанионная форма с обычным углеродом – три связи к азоту. По карбанионной форме отлично видим, что речь идёт о производных муравьиной кислоты, нитриле и амидинах. И об общей структурной особенности таких соединений – тот протон, который есть на углероде муравьиной кислоты, в таких производных муравьиной кислоты может быть оторван основаниями, а получающиеся карбанионы имеют карбеновую граничную структуру и некоторые особые свойства, присущие карбенам. Одна проблема – мы не очень хорошо понимаем какие.
Гетероциклические карбены (NHC)
Гетероциклические карбены тоже родились из цианид-иона. В конце 1950-х Рональд Бреслоу, один из самых знаменитых химиков 20 века, заметил аналогию между бензоиновой конденсацией и рядом ферментативных реакций, в которых альдегиды ведут себя тек, как будто они превращаются в некие интермедиаты с нуклеофильным карбонильным атомом.  Позже это явление назовут Umpolung. В этих ферментативных реакциях всегда участвует тиамин (витамин B1). Это не очень простая молекула, но Бреслоу предположил, что непосредственно в реакции участвует тиазольная часть этой молекулы, а точнее углерод между гетероатомами. С точки зрения этого углерода тиазолий представляет собой производное муравьиной кислоты (такие производные карбоновых кислот называют тиоамидами, а так как это такая форма, в которой двойная связь идёт к азоту, а простая – к сере, а не наоборот, то даже скорее изотиоамидами). Из обычной органической химии мы хорошо знаем, что производных у кислот много, а свойства у них в общем похожи. Бреслоу подтвердил свою гипотезу, сделав бензоиновую конденсацию с несколькими более простыми производными тиазолия, но не самого тиазола. Важно, чтобы атом азота был кватернизован, что делает его ещё более акцепторным. Тогда реакция идёт в очень мягких условиях в присутствии очень разбавленного водного раствора щёлочи. Это значит, что отщепление протона происходит очень легко, то есть соответствующий карбанион хорошо стабилизирован. Возьмём стандартный механизм бензоиновой конденсации и подставим в него вместо обычного цианид-иона карбанион из тиазолия, сократив его в Z.
Позже это явление назовут Umpolung. В этих ферментативных реакциях всегда участвует тиамин (витамин B1). Это не очень простая молекула, но Бреслоу предположил, что непосредственно в реакции участвует тиазольная часть этой молекулы, а точнее углерод между гетероатомами. С точки зрения этого углерода тиазолий представляет собой производное муравьиной кислоты (такие производные карбоновых кислот называют тиоамидами, а так как это такая форма, в которой двойная связь идёт к азоту, а простая – к сере, а не наоборот, то даже скорее изотиоамидами). Из обычной органической химии мы хорошо знаем, что производных у кислот много, а свойства у них в общем похожи. Бреслоу подтвердил свою гипотезу, сделав бензоиновую конденсацию с несколькими более простыми производными тиазолия, но не самого тиазола. Важно, чтобы атом азота был кватернизован, что делает его ещё более акцепторным. Тогда реакция идёт в очень мягких условиях в присутствии очень разбавленного водного раствора щёлочи. Это значит, что отщепление протона происходит очень легко, то есть соответствующий карбанион хорошо стабилизирован. Возьмём стандартный механизм бензоиновой конденсации и подставим в него вместо обычного цианид-иона карбанион из тиазолия, сократив его в Z.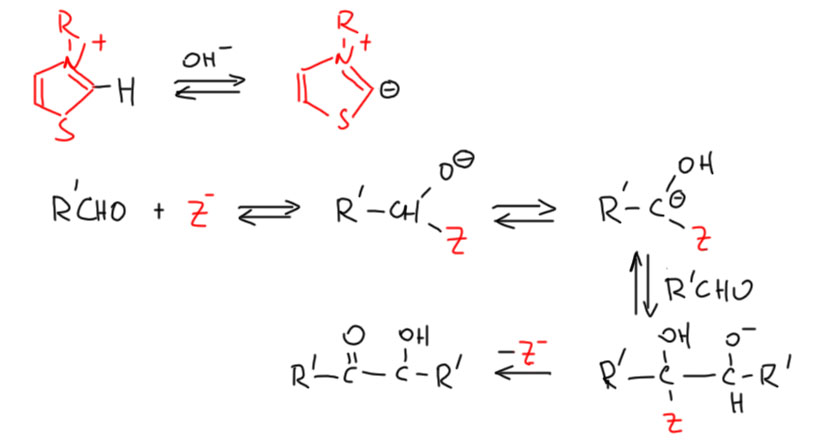
В этом месте возникает не очень простой вопрос. Если этот карбанион так хорошо стабилизирован, то что его стабилизирует? Простой индуктивный эффект соседних гетероатомов? Хорошо, но, видимо, мало, потому что далеко не все гетероциклы с такими атомами ведут себя похожим способом. Аналогия с цианид-ионом показалась Бреслоу вполне очевидной, и он и привёл структуру цианид-иона в виде мезомерии между обычной и карбеновой (цианидной и изоцианидной граничными структурами). Не могу не заметить, что в те времена этот инструмент был далеко не так востребован, как ныне, а в некоторых государствах даже и вовсе запрещён, как зловредное буржуазное извращение, не соответствующее учению марксизма-ленинизма.
Идея Бреслоу довольно быстро нашла развитие. Ханс-Вернер Ванцлик решил попробовать получить такой карбен, и проверить, не окажется ли он настолько стабилен, что его можно было бы выделить или, по крайней мере, как-то поймать. С “поймать” в те времена было не очень хорошо, поэтому расчитывали на типичную для карбенов реакцию, а такой и тогда и сейчас считается циклопропанирование. Перепробовав разные варианты, в том числе тиазолиевый, Ванцлик остановился на другом гетероцикле – имидазоле. Он, во-первых, симметричен, а это всегда проще, и во-вторых, азот более электроотрицателен чем сера, но и гораздо лучший мезомерный донор, чем сера. Депротонирование имдазолиевой соли должно дать карбанион, обладающий возможностью мезомерной стабилизации и имеющей две граничные формы – карбанионную и карбеновую, полностью аналогичные цианидной и изоцианидной формам цианид-иона.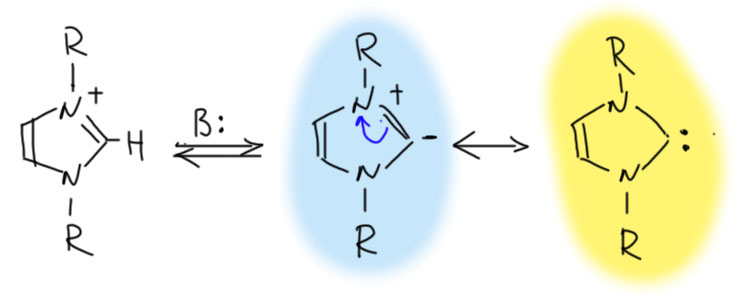 И имидазол – тоже производное муравьиной кислоты, амидин.
И имидазол – тоже производное муравьиной кислоты, амидин. После множества экспериментов с разными имидазолиевыми солями Ванцлик не смог получить то, что хотел, но увидел образование веьма необычного продукта – олефина, который можно представить как димер карбена. Ведь карбен – половинка олефина, значит, можно ожидать, что если мы получим карбен, он рекомбинирует в олефин, как радикалы рекомбинируют с образованием простой связи. Ванцлик даже предположил, что такая реакция может быть обратима, но не смог доказать это экспериментально. С тех пор равновесие карбен-олефин называют равновесием Ванцлика,
После множества экспериментов с разными имидазолиевыми солями Ванцлик не смог получить то, что хотел, но увидел образование веьма необычного продукта – олефина, который можно представить как димер карбена. Ведь карбен – половинка олефина, значит, можно ожидать, что если мы получим карбен, он рекомбинирует в олефин, как радикалы рекомбинируют с образованием простой связи. Ванцлик даже предположил, что такая реакция может быть обратима, но не смог доказать это экспериментально. С тех пор равновесие карбен-олефин называют равновесием Ванцлика,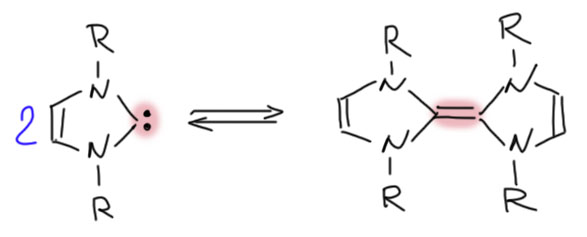 хотя так до сих пор и не удалось получить надёжных доказательств реальности такого равновесия, а образование олефина в эксперименте Ванцлика, как оказалось, объясняется совсем другой реакцией, но – с участием желаемого карбена. Иными словами, после Ванцлика удалось вполне точно доказать, что Ванцлик всё делал правильно и карбен у него был, но несовершенная техника эксперимента тех времён не позволила это доказать. Так или иначе, приоритет Ванцлика неоспорим.
хотя так до сих пор и не удалось получить надёжных доказательств реальности такого равновесия, а образование олефина в эксперименте Ванцлика, как оказалось, объясняется совсем другой реакцией, но – с участием желаемого карбена. Иными словами, после Ванцлика удалось вполне точно доказать, что Ванцлик всё делал правильно и карбен у него был, но несовершенная техника эксперимента тех времён не позволила это доказать. Так или иначе, приоритет Ванцлика неоспорим.
Но – проблема повисла и надолго. Всем показалось, что это экзотика, и к настоящей, большой химии отношения не имеет. Так, игрушка для любителей помучится, получить что-нибудь этакое, накропать красивую статейку, и прославиться. В большой же химии люди ищут серьезные способы синтеза и применяют их к важным, полезным молекулам. Никто к этим карбенам серьёзно не возвращался ло 1990-х, пока за дело не взялся американский химик Энтони Ардуэнго Третий. Ардуэнго с сотрудниками изучили работы Ванцлика, поняли, что он подошёл к решению проблемы вплотную, но немного не допёр, что нужно взять на азоте заместитель побольше чтобы помешать карбену димеризоваться в равновесии Ванцлика. Мелочиться не стали, взяли сразу адамантил. И получили, что хотели – стабильное кристаллическое вещество (Arduengo, A. J., III; Harlow, R. L.; Kline, M. A Stable
Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361), даже не требующее никаких особенных предосторожностей в работе кроме одного – оно не любит влаги. Воздух – пожалуйста, если сухой. И – отдельный признак очень стабильных, прямо таки дубовых веществ – высокая температура плавления без разложения (240ºС). 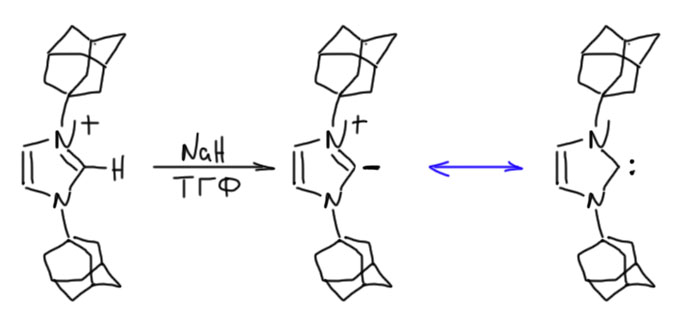
Ардуэнго привёл доказательства, что полученное им соединение – именно карбен. И это понятно, кому интересен ещё один карбанион, их к этому времени были известны многие тысячи. А вот стабильный карбен – это сенсация. Строго говоря, несколько стабильных карбенов до этого уже успели получить, но это были не самые простые соединения, да и стабильность их была не такой осязаемой, как у этого. Поскольку никаких реакций типических для карбенов с этим карбеном не получается, упирать пришлось на структуру. Самым важным и действительно поразительным свойством новой структуры было полное отсутствие признаков ароматичности. Исходный имидазолий – вполне ароматическая молекула с выровненными, насколько это возможно, длинами связей, максимально приближенная по форме к правильному пятиугольнику. А то, что получилось, было демонстративно уродливо – кольцо, конечно, оставалось плоским, но передний углерода выдвинулся вперёд, образовав валентный угол даже меньший, чем в sp3-гибридном состоянии. Меньший – это значит, что на этих связях увеличился p-характер, а поскольку, если в одном месте прибыло, то в другом должно убыть, а следовательно, орбиталь с неподелённой парой стала очень-очень похожа на почти чистую s-орбиталь, конечно, с небольшой примесью p, то есть, оставшись гибридной, стала очень компактной и низкой по энергии.
Посмотрим на эту ситуацию ещё раз, потому что она довольно необычна и в некотором смысле даже поразительна. Большинство из нас, если не вообще все без исключения живут с очень простой мыслью: ароматичность – это самое лучшее, что может случиться с молекулой. Если бы у молекул был рай, то там обитали бы исключительно ароматические молекулы, летали бы там в райском вакууме вместе со сферическими конями и пели осанну Творцу. Добровольно отказаться от ароматичности – нет, немыслимо! похоже на грехопадение, это карается изгнанием из рая, ну и так далее. Поэтому, когда нам рисуют молекулу, в которой можно, правильно расположив связи и правильно посчитав электроны, увидеть ароматичность, мы без колебаний так и поступим. Всегда, конечно, нужно правильно посчитать электроны, но в ароматичности, как и в выборах, всегда действует правило, – кто считает, тот и прав. Конечно, мы уже научились мириться с мыслью, что это касается только не очень больших циклов, и что, начиная с 8-членных циклов начинаются всякие капризы. Но уж для 5- и 6-членных циклов отказаться от ароматичности – немыслимое дело, сатанинские стихи.
Но вот если мы поверим Ардуэнго Третьему, то у него так и получается. Был ведь вполне ароматический имидазолий, и всё, что с ним сделали – просто оторвали протон из плоскости кольца – и получилась неароматическая частица. Вспомните опять обычную органическую химию – мы там много разбирали случаев, когда у ароматической молекулы что-то происходит в плоскости кольца, и всегда утверждали, что это никак не отражается на π-системе и её ароматическому характеру. Это так или мы что-то упустили?
На самом деле, и так, и не так. Подробнее здесь не будем, это долгий разговор, но ароматичность нельзя понимать как абсолютное благо. Это действительно благо (приводит к стабилизации молекулы), но относительное. В любой молекуле множество самых разных взаимодействий, стабилизирующих и дестабилизирующих, и реальная структура молекулы получается в результате нахождения компромисса, баланса между разными взаимодействиями. Ароматичность – только одно из них, хотя и очень весомое, но только в совершенно симметричных молекулах, с циклами, состоящими из одинаковых атомов (углерода чаще всего), это взаимодействие действительно имеет такой стабилизирующий вклад, что всё остальное почтительно расступается. В гетероциклах на это накладываются споры между атомами разной электроотрицательности, которым не очень хочется делить всё поровну, как и предполагает ароматическая делокализация. Мы отлично знаем, что, например, фуран имеет только эфемерные признаки ароматичности как раз потому что кислород не желает делиться парой электронов. Вот и в случае депротонированного имидазолия ситуация очень похожа. Посмотрим на граничные структуры этой молекулы. Их три, две одинаковые, соответствующие нашим представлениям от карбанионе из имидазолия. Мы считаем эти структуры ароматическими только потому, что они получена удалением протона из плоскости кольца изначально безусловно ароматического катиона имидазолия. Красным я показал пары и связи, участвующие в ароматической стабилизации. И третья структура – карбеновая. Мы в ней видим тоже непрерывный сопряжённый контур, где опять красным выделены участвующие в циклическом сопряжении (π-сопряжении) орбитали, но здесь это не только π-связь и пары, но и вакантная орбиталь на углероде (в химии нет общеупотребительного символа для пустой орбитали, как бы сказали в физике, дырки, поэтому используем просто пустой квадратик). Видим те же шесть электронов в цикле. Вывод простой – никто не лишает нас условий для ароматической делокализации ни в одной из этих структур. Вопрос только в том, насколько эта ароматическая делокализация важна для результирующей структуры. Ответ очевиден – не очень важна. Ароматическая стабилизация старается макксимально выровнять структуру – сделать максимально близкими по длине все связи, а цикл – максимально похожим на правильный пятиугольник. Другие факторы этому противостоят. Углерод предпочитает регибридизацию с тем, чтобы максимально понизить энергию орбитали, на которой находится пара. Его легко понять. Регибридизация неотвратимо приводит к увеличению длины связей C-N, а значит и с уменьшению эффективности взаимодействия на этом участке π-сопряжения, и к общему уменьшению стабилизации за счёт циклической делокализации. Азоты в этом случае тоже получают свой бонус – как более электроотрицательные атомы они получают свои пары в большее распоряжение (мы просто не должны забывать, что у структурах мы рисуем дискретные пары и связи, а в реальной молекуле каждый атом располагает своей, в общем случае нецелой частью электронной плоности), что снижает положительный заряд на этих атомах. В таком балансе взаимодействий и получается результирующая структура.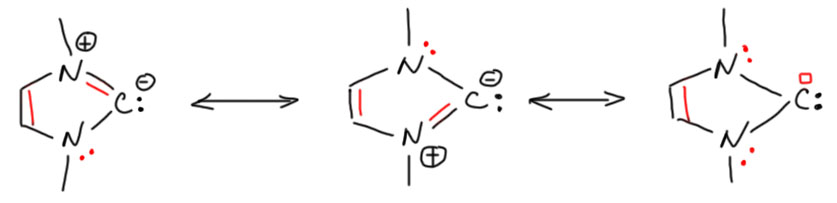
Подведём некоторые итоги. Можем ли мы считать, что карбеновая структура наилучшим образом отражает реальную структуру “карбена Ардуэнго”? Можем, но при условии того, что мы признаём и значительный вклад двух других структур. Является ли “карбен Ардуэнго” неароматическим? Да, но только в том смысле, что ароматическая делокализация, которая в этой молекуле обязательно есть (все три структуры имеют π-делокализацию и правильное число электронов в цикле) не даёт достаточного выигрыша энергии, чтобы обусловить образование максимально выровненной структуры, и стабилизация за счёт регибридизации, приводящая к частичному искажению геометрии кольца, выигрывает в балансе взаимодействий. Но баланс этот количественный (столько-то одного и столько-то другого), а не качественный (ароматическая – да или нет? а варианта “затрудняюсь ответить” нет). Из этого неоднозначного вывода следуют несколько важных вещей.
- для графического обозначения структры таких молекул после работ Ардуэнго и поныне предпочитают карбеновую структуру с простыми связями C-N, без зарядов на атомах, и с обозначением пары на углероде.
- когда заходит речь о реальных свойства таких молекул, невозможно не заметить их высокую основность – по разным оценкам карбены Ардуэнго относятся к самым основным органическим молекулам (имеются в виду нейтральные молекулы, а не карбанионы – не забывайте, что карбены Ардуэнго – незаряженные молекулы) с pK возможно до 30. Именно поэтому они так боятся влагу в растворителях и воздухе – просто протонируются обратно в банальные имидазолиевые соли. Но объяснить высокую основность карбеновой парой практически невозможно – приходится признать и немаленький вклад карбанионных граничных структур;
- те же структуры обусловливают высокую нуклеофильность “карбенов Ардуэнго” – на этой нуклеофильности основаны многочисленные применения этих карбенов в нуклеофильном органокатализе – увы, это выходит за рамки настоящего курса;
- когда из “карбенов Ардуэнго” образуются комплексы металлов, пара уходит на образование координационнной связи, а это сразу снижает выигрыш за счёт регибридизации и повышает вклад имидазолиевых граничных структур. В результате мы имеем одни из самых донорных лигандов в химии переходных металлов, практически не имеющих π-кислотности и не принимающих back-donation. Кстати, поэтому такие лиганды, хоть они и карбеновые, практически никогда не обозначают двойной связью металл-карбен. Связь всегда одинарная, координационная, в принципе точно такая же как в комплексах с триалкилфосфинами (у триарилфосфинов есть значительнай вклад back-donation), и поэтому NHC-лиганды в комплексах часто замещают триалкилфосфины и играют ту же роль. Такие лиганды стабилизируют более высокие степени окисления, а металлы делают более нуклеофильными – им же надо куда-то девать свою плотность, а этот лиганд её не принимает – значит она пойдёт в другие лиганды, например, те, которые участвуют в реакции.
“Насыщенные” и “ненасыщенные” гетероциклические карбены
Так как Ардуэнго пришёл к выводу, что полученные им карбены – это именно карбены, большой необходимости в том, чтобы иметь ароматическое исходное, имидазолиевую соль, нет. И можно взять неароматическое исходное, имидазолин (имидазолин – это дигидроимидазол, с одной двойной связью, а совсем насыщенный называется имидазолидин, но он бесполезен для получения карбена), тоже кватернизованный, отнять у него протон и получить стабильный карбен. Эта гипотеза вполне сработала. Первый такой карбен имел два объёмистых мезитильных заместителя на азотах.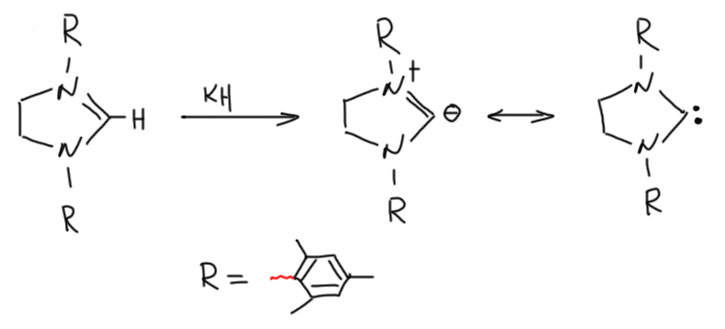
Прежде чем двигаться дальше, введём систематические названия. Карбен обозначают окончанием -илиден (-ylidene). Поэтому карбен из имидазолия называют имидазол-2-илиден (imidazol-2-ylidene). Из имидазолиния – имидазолин-2-илиден. Номера у азотов – 1 и 3. 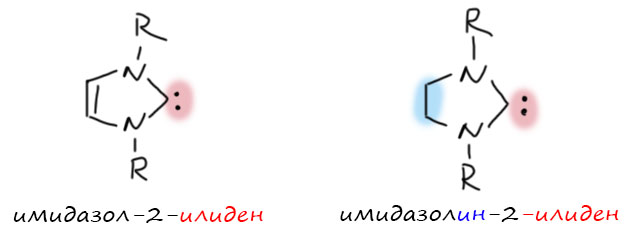
Итак, карбен получился такой же, но не совсем. Стабильный, но не настолько, как такой же, но из имидазолия. Но это мелочи. Важнее то, что он ещё более донорный, и как лиганд, и как основание. Причина этого проста – мы всё же не можем в имидазолиевом карбене игнорировать ароматическую делокализацию, частично оттягивающую электронную плотность от карбенового углерода. В “насыщенном” аналоге этого эффекта нет.
В результате всех этих изысканий сложился набор из четырёх карбенов Ардуэнго, наиболее часто используемый в исследованиях и применениях. Сложились и сокращения. Во-первых, всек карбены Ардуэнго (честнее было бы их называть карбенами Ванцлика-Ардуэнго) и их многочисленные аналоги стали называть тремя буквами – NHC. Это значит N-гетероциклические карбены (N-heterocyclic carbenes). Вообще-то за последние 20 лет этих стабильных карбенов наполучали сотни, и далеко не все из них происходят от азотсодержащих гетероциклов. Но большинство происходят, поэтому решили не париться и использовать эти 3 буквы почти всегда, хотя бы если в основе карбена есть какой-то гетероцикл.
Набор из четырёх самых популярных NHC состоит из двух пар. На азоте используют два заметителя – объёмистый мезитил и ещё более объёмистый 2,6-диизопропилфенил. И каждая пара состоит из имидазолиевого и имидазолинового карбена. Получаем множество, в котором можем увеличивать стерический объём и донорность. Называют их очень просто – имидазольный остов сокращают в I, насыщенный в SI. И добавляют заместитель. Вот эта священная четвёрка, с которой вы будете в современной химии сталкиваться гораздо чаще, чем с трифенилфосфином. 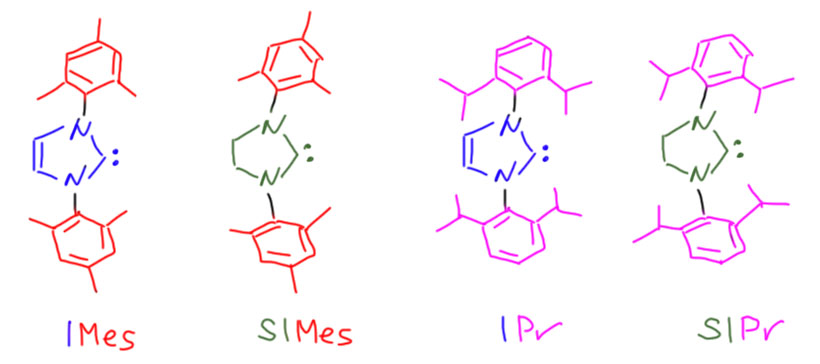
Иногда, редко, но бывает так, что требуется ещё больший стерический объём. Тогда вспоминают про самый первый карбен, полученный Ардуэнго, называя его по той же схеме IAd. И к нему тоже есть пара SIAd.