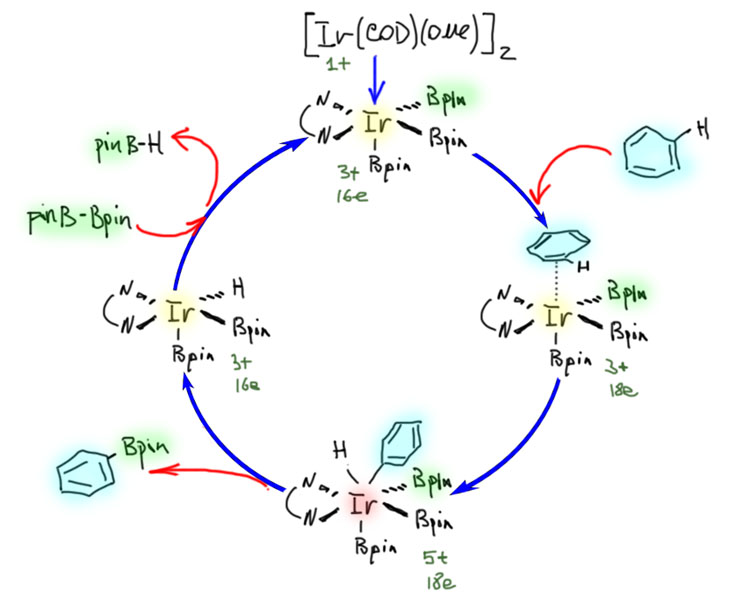
CH-активация
Реакции органических соединений чаще всего происходят на определенных реакционных центрах, атомах, несущих уходящие группы, включенных в кратные связи и т.п. Но больше всего в органических соединениях атомов водорода, связанных с атомами углерода. Это настолько банальная и вездесущая часть структур органических соединений, что мы обычно вообще их не замечаем, никак не обозначаем их в структурных формулах, – просто и совершенно инстинктивно мысленно добавляем атомы водорода для соблюдения правильных валентностей.
Тем не менее, именно атомы водорода на углеродах – самые лакомые кусочки органической структуры для органиков, разрабатывающих новые методы синтеза. Если удается целенаправленно и более-менее селективно заменить атом водорода на какую-нибудь группу или фрагмент, радости не бывает пределов, и самые гламурные научные журналы с радостью распахивают свои страницы для таких сообщений и статей. Почему? Строго говоря, потому, что это во-первых очень модно, и все тут, а про во-вторых лучше не спрашивать. Это главное. Научная жизнь в целом устроена точно так же, как и жизнь мира моды и шоу-бизнеса. Объяснить, почему что-то сейчас в тренде, а что-то украсило собой задворки жизни, практически невозможно.
Но наука, безусловно, всегда старается для любого тренда придумать рациональное обоснование. В случае с атомами водорода в ход идет, в первую очередь, спасительный принцип экономии атомов. По крайней мере, с точки зрения исходных веществ и продуктов замена атомов водорода на что-то важное и полезное более всего удовлетворяет этому принципу из всех реакций замещения (реакции присоединения в любом случае вне конкуренции) – так как замещается, уходит в помойку, самый легкий из всех известных атомов, атом водорода. В этом месте обязательно нужно сделать вид, что мы не знаем, что атомы водорода сами по себе на помойку не отправляются, а непременно прихватывают еще что-нибудь – кислотный остаток, основание, акцептор атома водорода, из того, что было в реакционной смеси помимо исходного органического субстрата, и масса этого довеска будет в десятки, сотни, а то и тысячи раз превосходить массу атома водорода. Но умение деликатно делать вид, что мы не замечаем что-то такое, что замечать в данном контексте не положено – важнейший признак образованного, культурного человека. Так же и в мире моды – мы же не спрашиваем, отчего вон та модель надела на голову ведро. Если пришли на модный показ, подразумевается, что знаете, отчего можно надеть на голову ведро. А если не знаете и позволяете себе мерзко хихикать – прочь с модного показа, невежда!
Но если еще немного подумать, то придется признать, что доля правды в представлении об “атомной экономичности” методов, связанных с CH-активацией, все же есть. Если в начале всех синтетических цепочек стоят самые простые исходные, то для того чтобы ввести в молекулы хорошие уходящие группы или другие реакционные центры приходится использовать всякие вспомогательные реакции (галогенирование, нуклеофильное замещение и т.п.), часто с введением и снятием защитных групп, и цена превращения сильно возрастает, а количество отходов увеличивается. Тогда и правда проще и экономичнее придумать метод прямой модификации исходных по CH-связям. Реальность всегда сложнее принципов и идей, поэтому примем простую мысль, что запас карман не тянет, и лишние методы никогда не помешают. В лабораторном синтезе, да и в тонком органическом синтезе ценных соединений возможность получить продукт всегда важнее пути, которым этот синтез осуществляется.
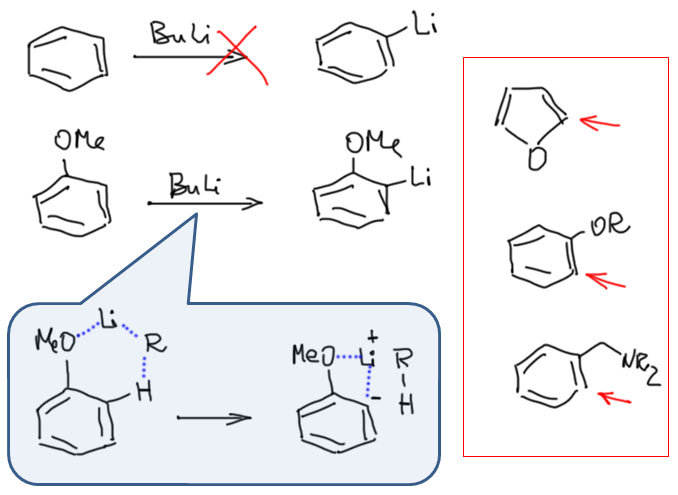
Итак, мы хотим использовать в реакциях в качестве реакционных центров самые обычные CH-связи. Подразумевается, что обычно они недоступны или труднодоступны для реакций. Дело здесь часто бывает вовсе не в низкой реакционной способности таких связей, а скорее в том, что в органических молекулах полно разных связей этого типа, и задача целенаправленно выбрать только какие-то конкретные может на первый взгляд показать ся вообще нерешаемой. Но она решается, и весьма типичным для органической химии способом. Нужно найти побольше методов, позволяющих воздействовать на связи нужного типа. У каждого из таких методов будут свои требования к структуре, электронным эффектам, влиянию соседних групп, стерическим факторам. Если мы располагаем большим набором методов, то в том случае, когда нам понадобится реально добраться до какого-то конкретного атома водорода в конкретной молекуле, есть надежда, что минимум один из коллекции методов подойдет. И тогда мы прославим подходящих богов, и скажем, что именно так и было задумано.
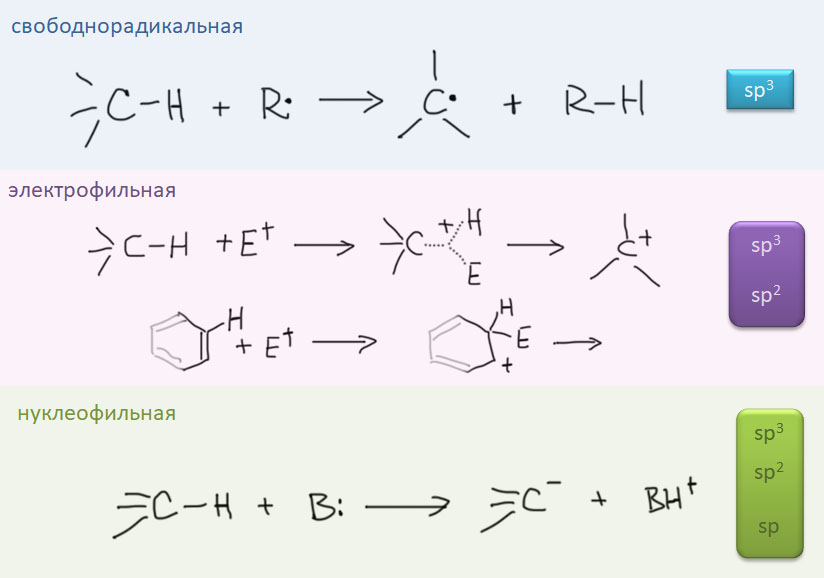
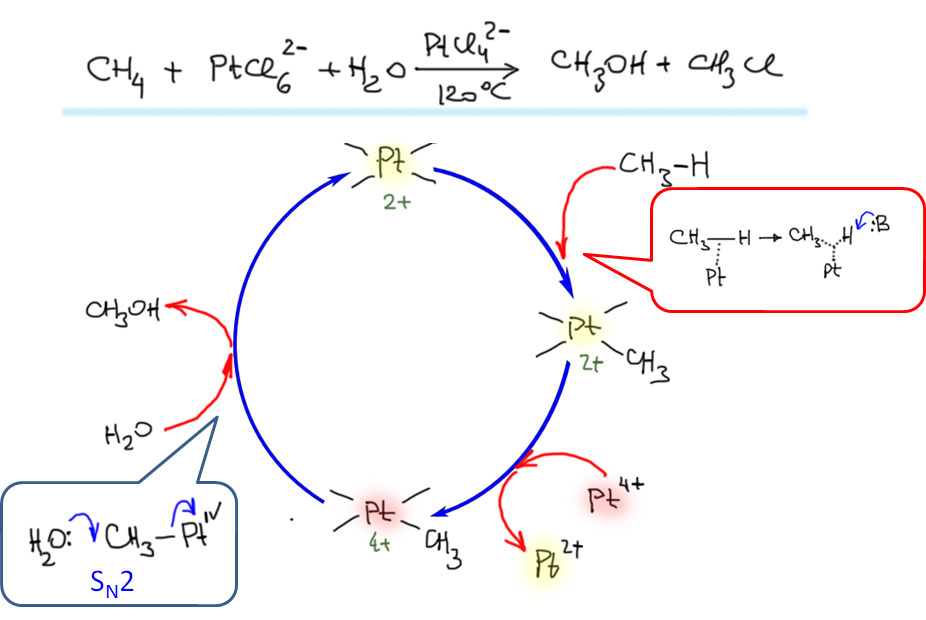
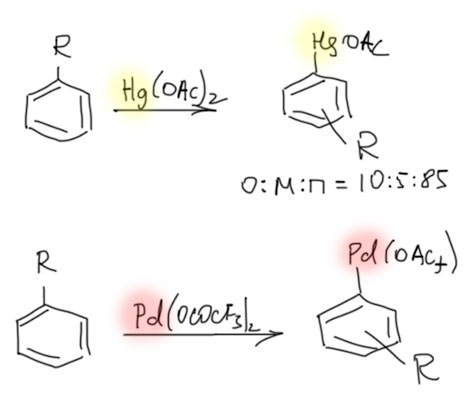
Реакция Фудзивары-Моритани
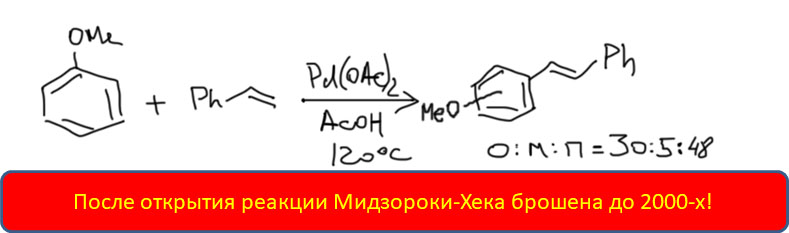
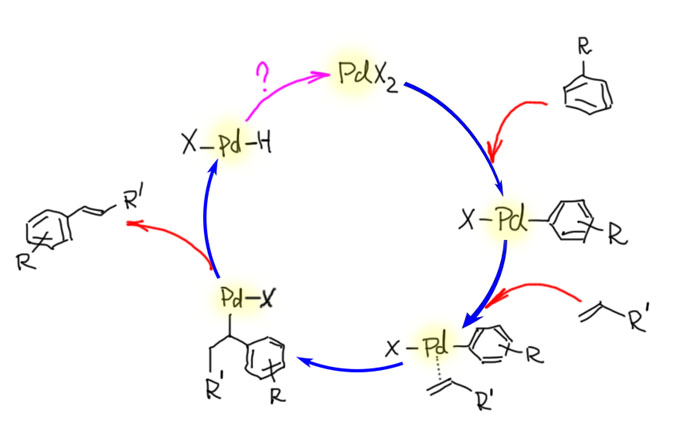
Первое реальное применение реакции электрофильного палладирования было предложено Фудзиварой и Моритани в 1967 году, на год раньше реакции Мидзороки-Хека, – обе реакции являются ближайшими аналогами. В отличие от реакции Мидзороки-Хека, в которой всегда образуется конкретный продукт, в реакции Фудзивары-Моритани региоселективность определяется обычными правилами ориентации в ароматическом электрофильном замещении, а следовательно получается смесь продуктов. Низкая селективность реакции Фудзивары-Моритани обусловила почти полное забвение этой реакции до начала 2000-х.
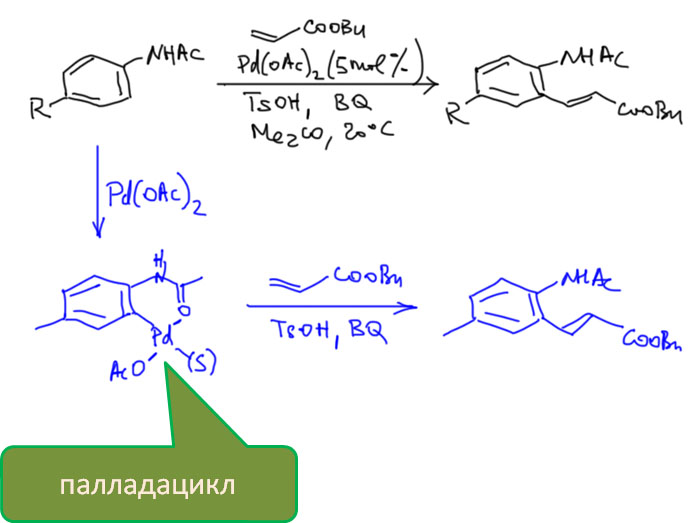
Палладациклы
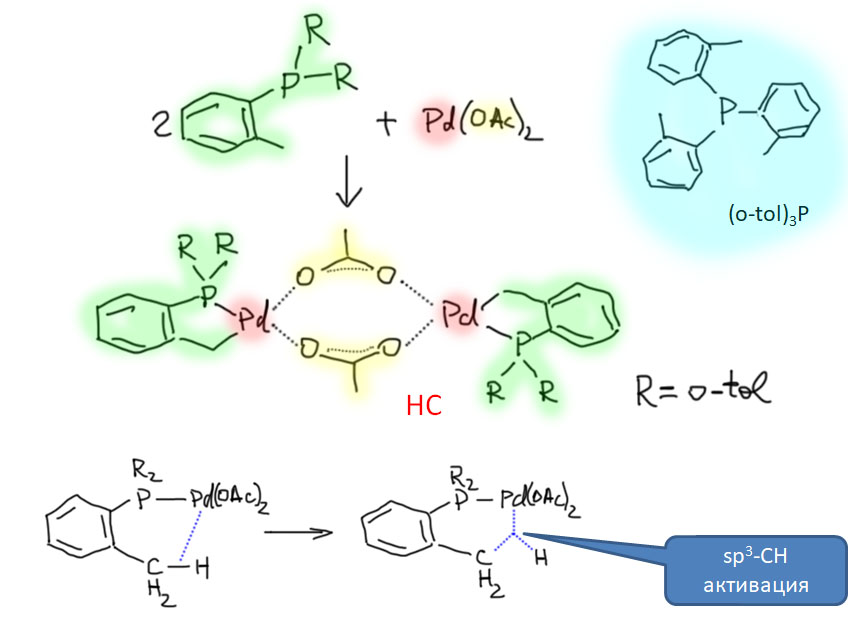
Пяти- и шестичленные палладациклы очень легко получаются из самых разных ароматических соединений, если в их составе есть направляющая группа или фрагмент. Легкодоступность палладациклов в проклятые 90-е годы сыграла злую шутку с большим количеством исследователей, потому что в них стали видеть просто все на свете, включая новый класс катализаторов. Дальнейшие исследования внесли ясность в эту проблему, и выявили реальные ценные свойства этих металлоорганических соединений.
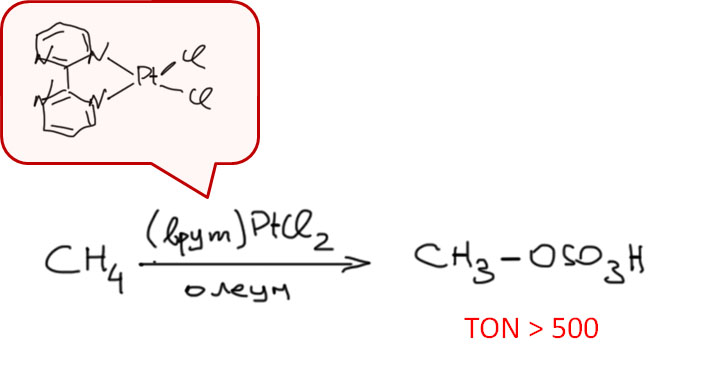
Огромные величины TON
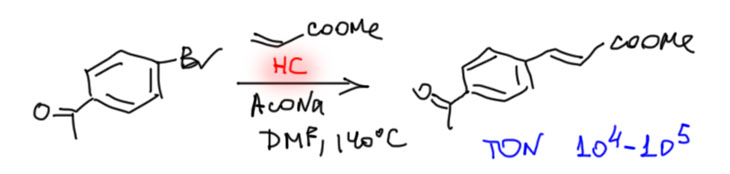
можно получить в реакциях Мидзороки-Хека и кросс-сочетания по Судзуки-Мияура в присутствии палладациклов, но реакции ограничены субстратами с высокой реакционной способностью и требуют весьма жестких условий.
Пинцерные палладациклы
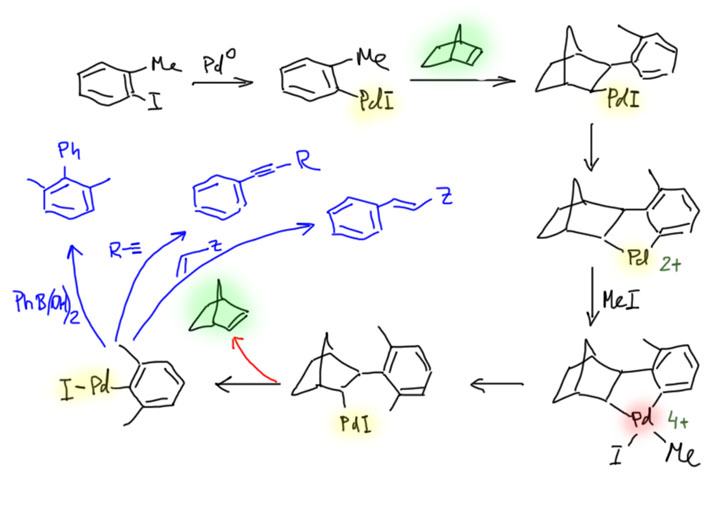
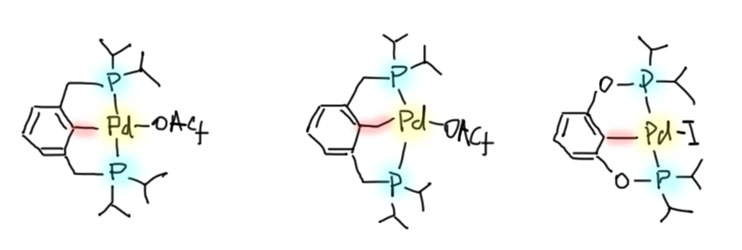 Охота за все боле и более активными (или кажущимися таковыми) палладациклами довольно быстро привела к так называемым пинцерным палладациклам, то есть комплексами с тридентатными лигандами, минимум одно из координационных центров в которых образуется за счет внутримолекулярного направленного палладирования. С такими комплексам были получены TON, превышающие миллион циклов. Но именно в момент этого триумфа возникли первые сомнения в том, что объяснение таких результатов выдающейся устойчивостью таких систем (напомню, что TON – это мера долговечности каталитической системы в условиях реакции) имеет право на существование. Проблема в том, что если эти комплексы действительно так устойчивы, то на атоме палладия просто не хватает свободных координационным мест даже для кросс-сочетания, а тем более для реакции Мидзороки-Хека. К тому же никто и никогда не мог показать, что палладий в палладацикле может быть восстановлен до нульвалентного состояния без разложения комплекса.
Охота за все боле и более активными (или кажущимися таковыми) палладациклами довольно быстро привела к так называемым пинцерным палладациклам, то есть комплексами с тридентатными лигандами, минимум одно из координационных центров в которых образуется за счет внутримолекулярного направленного палладирования. С такими комплексам были получены TON, превышающие миллион циклов. Но именно в момент этого триумфа возникли первые сомнения в том, что объяснение таких результатов выдающейся устойчивостью таких систем (напомню, что TON – это мера долговечности каталитической системы в условиях реакции) имеет право на существование. Проблема в том, что если эти комплексы действительно так устойчивы, то на атоме палладия просто не хватает свободных координационным мест даже для кросс-сочетания, а тем более для реакции Мидзороки-Хека. К тому же никто и никогда не мог показать, что палладий в палладацикле может быть восстановлен до нульвалентного состояния без разложения комплекса.
Пинцерные металлациклы и комплексы
Жесткая структура пинцерных металлациклов не дает им участвовать в реакциях кросс-сочетания, Мидзороки-Хека и т.п. без разрушения, но может быть очень полезной в других реакциях, где остающихся координационных возможностей достаточно, а жесткий контроль координационной сферы ограничивает число возможных путей превращения.
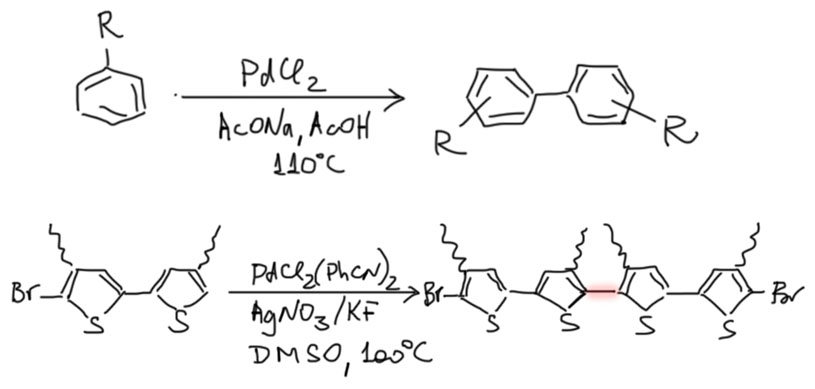
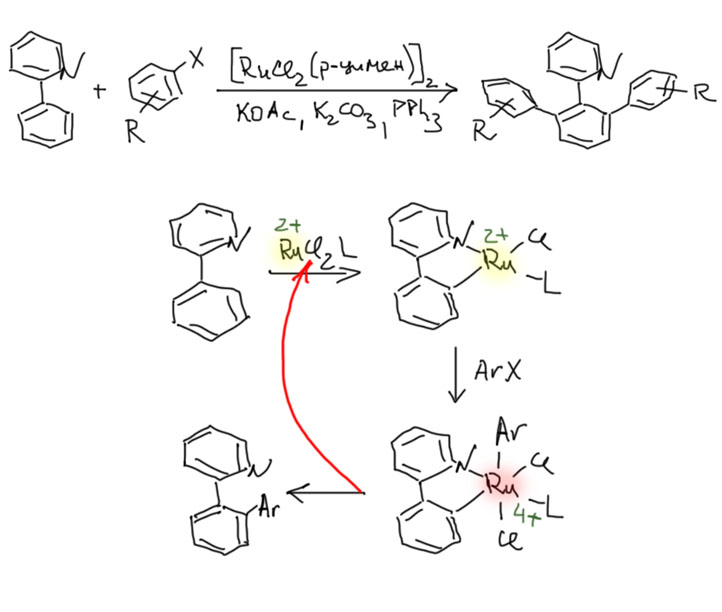
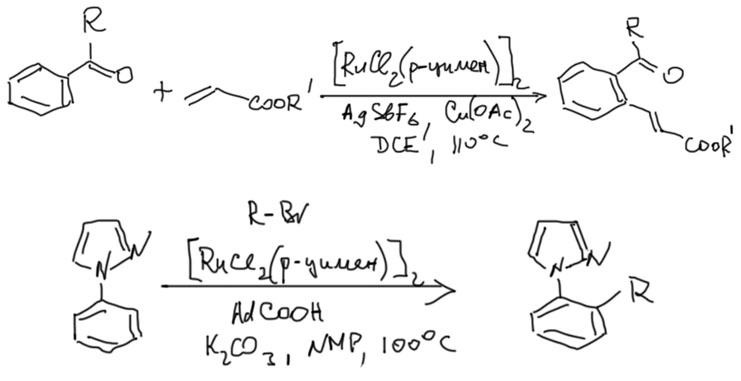
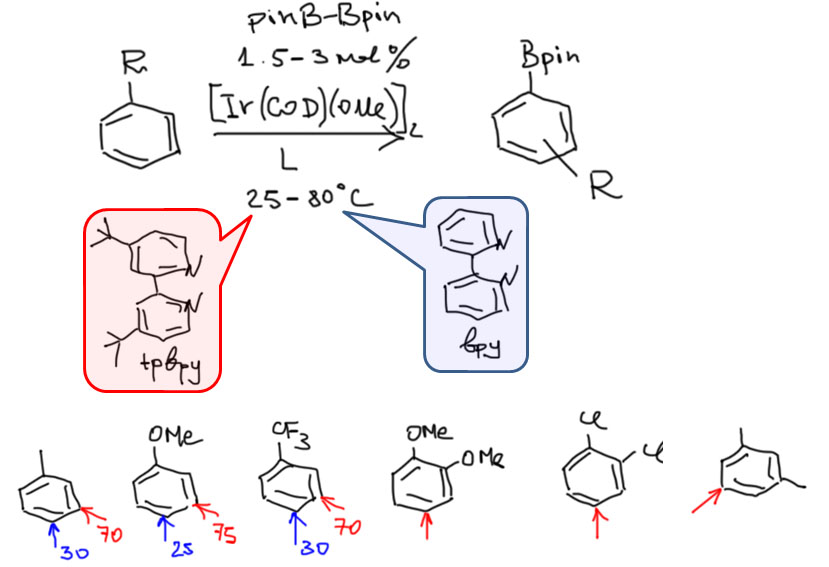
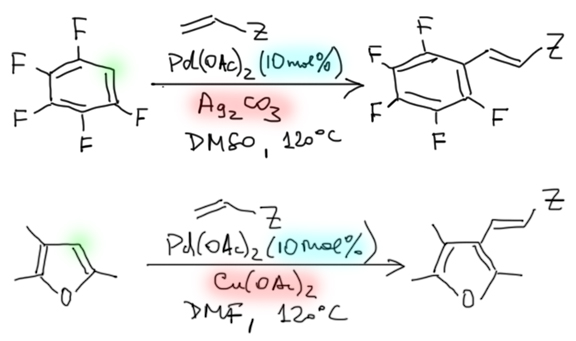
Накопление данных по CH-активации привело к невероятно бурному и быстрому росту числа публикаций, описывающих методы так называемого прямого арилирования, то есть фактически кросс-сочетания между ароматическими соединениями за счет C-H связей и без необходимости введения уходящих групп. Конечно, все такие методы драматически уступают классическому кросс-сочетанию в широте – каждый протокол прямого арилирования обычно очень узок и применяется только к субстратам определенного типа, а любой шаг влево или шаг вправо от субстратов, описанных в очередной статье, оказываются шагом в никуда. тем не менее, это очень удобно, и методы стали реально применять в синтезах. В настоящее время в этой области наметилось три металла с максимальным потенциалом, и это вся благородная тройка из второго ряда – рутений, родий, палладий. Бросим торопливый взгляд на достижения этой компании.